
电化学法处理香料工业氯化钠有机废盐的研究
2024-03-25 09:10姚健康胡硕真钮东方吴建平张新胜
无机盐工业 2024年3期
姚健康,胡硕真,钮东方,吴建平,张新胜
(1.华东理工大学化学工程联合国家重点实验室,上海 200237;2.上海瑞元化学科技有限公司,上海 200231)
香料是重要的食品添加剂之一,其种类有5 000多种,每年全球的香料需求增速达到5.4%,因此其工业生产中产生的废盐也越来越多[1-2]。这些废盐中通常含有大量具有毒害性的有机物,因此不能被直接当作工业原料再利用[3],如果不经过无害化处理就被随意处置,不仅会对生态环境造成严重污染,还会导致盐类资源的大量浪费[4-7]。
当前,绝大部分工业废盐的处理方式为企业建库暂存或直接填埋,这种方式虽然简单且成本低,但对周围环境有明显的二次污染隐患,而对于废盐中的盐分,通过合理的处理手段仍然具有资源化利用价值。工业废盐的无害化和资源化处理方法主要包括物理法、热法、模拟废水法、化学转化法等。物理法主要包括洗盐法[8]和萃取法[9]等,这种方法操作简单但存在二次污染;热法主要包括热解碳化[10-13]和高温熔融法[14-15],通过高温加热去除废盐中的有机物,该方法对有机物去除率高、反应时间快,但存在尾气污染等问题;模拟废水法则是利用废盐的高溶解性将废盐溶解为高盐有机废水,再利用高盐有机废水领域的处理方法对废盐进行无害化和资源化处理,该类方法主要包括生物法[16-17]、膜法[18-19]和高级氧化法[20-24]等;化学转化法是将废盐中的盐分通过化学法转化为价值更高的工业产品,主要包括氯化钠废盐制纯碱[25]和硫酸钠废盐制硫酸钾[26]等。
氯化钠有机废盐溶解后制成的高盐有机废水具有高电导率的特性,电化学技术在该类废水处理中具有独特的优势,这方面的研究也得到人们越来越多的关注[27-28]。电化学法具有工艺流程简单、反应条件温和、无投料、无二次污染及反应易控制、绿色环保等优点,具有很强的发展潜力和工业应用前景。本研究采用电化学氧化耦合膜法的一膜两室电解工艺在降解废盐中毒害性有机物的同时制取氢氧化钠,为该类废盐的资源化处理提供参考。
1 实验部分
1.1 仪器与试剂
单室电解槽(实验室自制);双室电解槽(实验室自制);UTP1306S型直流稳压稳流电源;DF-101S型恒温磁力水浴锅;COD-571 型COD 测量仪;COD-571-1型消解仪;PHSJ-3F型pH计。
硫酸(纯度95%~98%)、盐酸(纯度36%~38%);重铬酸钾、硫酸银、硫酸汞、硝酸银、氯化钾、铬酸钾、氢氧化钠、硝酸、硝酸钠、碳酸钠、酚酞、N,N-二乙基-1,4苯二胺(DPD)、硫酸亚铁铵,均为分析纯;邻苯二甲酸氢钾为优级纯;去离子水由实验室自制。
1.2 模拟废水基本信息
表1为模拟废水基本信息。

表1 模拟废水基本信息Table 1 Basic information of simulated wastewater
1.3 单室电化学氧化实验
在单室电解槽中,以Ti/PbO2电极(有效面积为26 cm2)为阳极,不锈钢电极(有效面积为26 cm2)为阴极。量取400 mL 模拟废水为反应液加入电解槽中,控制阴、阳极板间距,使用HCl 和NaOH 调节反应液初始pH,用恒温磁力水浴锅将反应液加热至目标温度并保持恒温通电反应。保持电流恒定电解8 h,每隔2 h取样并记录反应液体积。
1.4 一膜两室电解实验
在双室电解槽中阳极液为300 mL模拟废水,阴极液为250 mL氢氧化钠溶液,阳离子交换膜为DF988(山东东岳),实验过程与单室电化学氧化实验一致。
1.5 实验分析方法和计算公式
1.5.1 化学需氧量的检测方法
实验所得的样品,使用银量法测定其Cl-含量,并用去离子水将其稀释至Cl-质量浓度小于1 000 mg/L后待测。取2 mL 待测液加入3 mL 氧化剂(含质量分数为1%硫酸银的硫酸与0.009 mol/L 的重铬酸钾溶液按体积比2∶1混合),150 ℃下消解2 h后得到消解液,将该消解液冷却至室温后,使用化学需氧量(COD)测量仪进行样品COD的测量。
1.5.2 稀释倍数法分析色度
1)准确移取10 mL 澄清试样于100 mL 容量瓶中,用去离子水稀释至100 mL。混合均匀后在自然光源充足的条件下,将稀释后的试料和去离子水分别倒入50 mL 具塞比色管至50 mL 标线,将具塞比色管垂直放置在白色表面上,垂直向下观察液柱,比较试料和去离子水的颜色。如果还有颜色,则继续取稀释后的试料10 mL,再稀释10 倍,依次类推,直到刚好与去离子水无法区别为止,记录稀释次数n。
2)用量筒取25 mL步骤1第n-1次稀释的试料,从小到大逐级按自然倍数进行稀释,每稀释一次,混匀后按与步骤1 相同的方法观察,直到刚好与去离子水无法区别时停止稀释,记录稀释倍数D1。样品的色度(D),按式(1)进行计算:
1.5.3 活性氯的检测
活性氯采用HJ585-2010《水质 游离氯和总氯的测定N,N-二乙基-1,4-苯二胺滴定法》的N,N-二乙基-1,4苯二胺(DPD)滴定法进行测定。
在250 mL 锥形瓶中依次加入15 mL 磷酸盐缓冲液、5 mL DPD 溶液和体积为V0的待测水样,均匀混合后立即用硫酸亚铁铵标准滴定液滴定至无色为终点,滴定液消耗体积为V1。计算公式如下所示:
式中:C为硫酸亚铁铵标准滴定液浓度,mmol/L;70.91 为Cl2的相对分子质量;2 为每1 mol 硫酸亚铁铵相当于氯(Cl2)的物质的量倍数;C(Cl2)为Cl2的浓度,mmol/L。
1.5.4 氢氧化钠浓度测定
使用酸碱中和滴定法对电解过程中产生的氢氧化钠浓度进行测定。以无色酚酞溶液为指示剂,0.05 mol/L的H2SO4标准溶液对待测NaOH样品进行滴定。NaOH浓度计算公式如下:
式中:CNaOH为物质的量浓度,mol/L;V为NaOH 样品体积,mL;VH2SO4为消耗H2SO4标准溶液体积,mL。
1.5.5 计算公式
实验中各项指标:废水COD去除率Y、COD降解电流效率η和氢氧化钠的生产电耗E的计算方法如下:
式中:Y废水COD 去除率,%;COD0为电解液初始COD值,mg/L;CODt为t时刻电解液COD值,mg/L;V0为电解液初始体积,L;Vt为电解液t时体积,L;F为法拉第常数,96 485 C/mol;z为生成一个氧原子转移的电荷数,此处为2;I为电流,A;t为反应时间,s;E为氢氧化钠的生产电耗,kW·h/kg;为氢氧化钠初始浓度,mol/L;CtNaOH为氢氧化钠t时刻浓度,mol/L;U为槽电压,V 。
2 结果与讨论
2.1 电化学氧化实验条件的研究
2.1.1 电流密度对有机物降解效果的影响
电流密度对电化学反应速率、反应时间及反应程度都有很大影响,是影响电化学氧化过程的重要因素,因此选择合适的电流密度对电化学氧化过程十分重要。本节在电解温度为35 ℃,不改变废水初始pH(pH为6)的条件下,探究电流密度对废水有机物降解的影响。
图1为不同电流密度下废水COD去除率随电解时间的变化曲线。从图1 可以看出,在实验电流密度范围内,废水COD去除率随电流密度的增加而增加。电解8 h且当电流密度上升至600 A/m2时,废水COD 去除率可达92.3%。而当电流密度由600 A/m2进一步增加至700 A/m2时,反应前期COD 的去除率仍然有所提高,但是随着电解时间的增加,其COD的去除率与电流密度为600 A/m2时基本一致,此时提高电流密度已难以达到提高COD 去除率的效果。

图1 同电流密度下废水COD去除率随时间的变化情况Fig.1 Changes of removal rate of wastewater COD with time at different current densities
当电流密度提升至600 A/m2和700 A/m2时,电解6 h 后废水COD 去除率达到85%左右,COD 浓度降至约为480 mg/L。由于有机物浓度较低,即使进一步增加电流密度来提高电极上氧化活性物质的生成速率,这些氧化活性物质(羟基自由基等)的利用率也难以保证,更多的羟基自由基(·OH)参与析氧副反应[29],难以有效地增加有机物的降解速率。因此,当电流密度大于600 A/m2后难以进一步有效提高COD去除率。
不同电流密度下,电解8 h 后的COD 降解的电流效率如表2 所示。从表2 可以看出,电解8 h 的COD降解的电流效率随着电流密度的增加而降低,电流效率由300 A/m2的49.2%下降至700 A/m2的28.0%。相同电解时间下,电流密度越大有机物降解量越多,阳极液有机物含量越低,阳极产生的氧化活性物质的利用率及有机物与阳极接触发生直接氧化的概率降低,因此,电流效率随电流密度的增加而降低。

表2 电解8 h后不同电流密度下COD降解电流效率Table 2 Current efficiency of COD degradation at different current densities after 8 h of electrolysis
图2和表3分别为不同电流密度下废水COD降解过程的动力学拟合曲线和动力学参数。由图2可知,不同电流密度下电化学氧化降低废水COD的过程均符合一级动力学模型,且反应速率随着电流密度的增加而增加,这与文献报道一致[30]。由表3 可知,反应速率常数k随着电流密度的增加而增加,从电流密度为300 A/m2下的0.133 h-1增加到电流密度为700 A/m2下的 0.328 h-1。各动力学拟合曲线的决定系数R2值均大于0.98,拟合曲线的决定系数R2越接近1表示拟合度越高,因此图2所示的各曲线均有良好的拟合度。

图2 不同电流密度下COD降解动力学曲线的变化情况Fig.2 Change of COD degradation kinetics curves at different current densities

表3 不同电流密度下废水COD降解动力学参数Table 3 Kinetic parameters of COD degradation in wastewater at different current densities
综合电流效率、COD去除率等因素,确定600 A/m2为电化学氧化降解该废水中有机物的最佳电流密度。
2.1.2 反应温度对有机物降解效果的影响
电化学氧化降解有机物的过程中,温度对氧化反应速率和分子扩散等都有很大的影响。因此,本节在电流密度为600 A/m2,不改变废水初始pH(pH为6)的条件下,探究反应温度对电化学氧化降解废水有机物的影响。
图3为不同反应温度下COD去除率与动力学拟合曲线的变化情况。由图3 可知,随着反应温度的增加废水COD 去除率和去除速率均呈现先增加后降低的趋势。在电解8 h、35 ℃时废水COD 去除率最大(92.3%)。
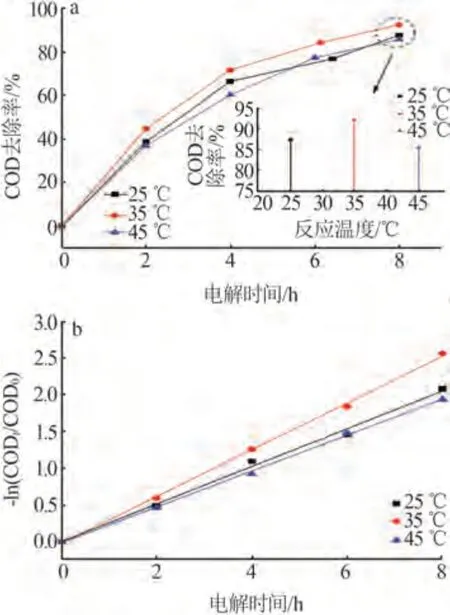
图3 不同温度下COD去除率(a)和动力学曲线(b)的变化情况Fig.3 Change of COD removal rate(a) and degradation kinetics curves(b) at different temperatures
当反应温度由25 ℃提升至35 ℃时,不仅加快了有机物在溶液中的扩散传质,使其更易扩散至电极表面被直接或间接氧化降解,还增加了有机物与氧化活性物质的氧化反应速率,使得COD去除率和去除速率提高。
表4 为电解2 h 时废水中活性氯含量随温度的变化情况。从表4可以看出,2 h时废水中活性氯含量随着温度的增加而不断降低。这可能是因为温度的升高使得活性氯稳定性和Cl2的溶解度降低[31]。

表4 电解2 h后废水中活性氯含量随温度的变化情况Table 4 Change of active chlorine content in wastewater with temperatures after 2 h of electrolysis
当反应温度上升至45 ℃时,虽然可以进一步加快物质扩散和有机物氧化反应速率,但废水中活性氯含量较温度为25 ℃和35 ℃时有大幅度降低,这导致反应过程中活性氯对有机物的间接氧化反应减少,有机物的降解效率降低。同时,宋红等[32]研究表明,电极的析氧过电位随着反应温度的升高而降低,即高温利于析氧副反应的发生,电极生成的·OH 更多的参与到析氧反应,这导致了45 ℃时COD去除率和去除速率的降低。
综上所述,反应温度过低不利于反应体系中有机物扩散传质和氧化反应速率的提升,而温度过高不利于氧化活性物质(活性氯和·OH 等)的生成,因此在所选温度内35 ℃为电化学氧化降解该废水中有机物的最佳反应温度。
2.1.3 初始pH对有机物降解效果的影响
pH 是电化学氧化降解有机物过程中一个重要的影响因素,大量研究表明,溶液pH对反应电位、有机物和活性氧化物质的存在形式及活性氧化物质的氧化能力等方面都有非常大的影响。因此,本节利用HCl和NaOH将模拟废水原液的初始pH由6分别调至3、9和12后,在电流密度为600 A/m2,反应温度为35 ℃的条件下,探究废水初始pH(pH0)对电化学氧化降解废水中有机物的影响。
图4 为不同初始pH 下废水COD 去除率随电解时间的变化曲线。从图4可以看出,废水pH0为9和12时,电解前期的COD去除率显著降低。虽然整体上废水COD 去除率随着废水pH0的增加而降低,但是各组废水COD去除率之间的差距也明显缩小,具体COD去除率数值如表5所示。
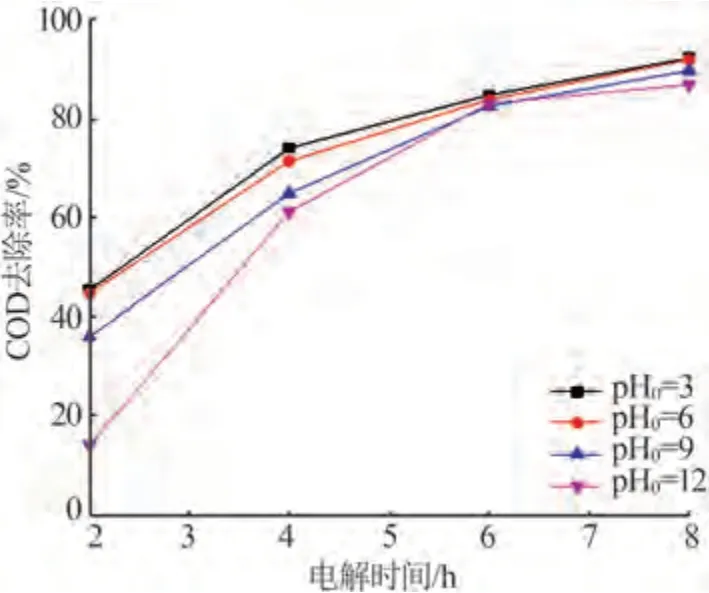
图4 不同废水初始pH下废水COD去除率随时间变化情况Fig.4 Change of COD removal rate of waste water with time at different initial pH

表5 各组实验的废水COD去除率Table 5 COD removal rate of each group of experiments
顾坤[33]研究表明,在碱性条件下阳极析氯过电位增加,会导致活性氯生成量降低。同时在碱性条件下,·OH氧化能力也会下降[34-36],这些都使得废水有机物氧化速率降低。因此废水pH0为9 和12 时,电解前2 h的COD去除率较低。
随着电解的进行,废水pH 会有所变化,图5 为废水pH随电解时间变化的曲线。从图5可以看出,在电解4 h 后,不同初始pH 下的废水pH 趋于接近,因此有机物的电化学氧化条件基本一致,废水中有机物含量越充足则氧化活性物利用率越高,其COD降解速率越大。

图5 不同初始pH下废水pH随时间的变化情况Fig.5 Change of wastewater pH with time at different initial pH
前4 h的电解过程中,pH0为9和12的模拟废水COD 去除率较低,废水中COD 含量高。因此,电解4 h后,在有机物的电化学氧化条件基本一致的情况下,pH0为9和12的模拟废水COD降解速率加快,各组实验在反应6 h 和8 h 后COD 去除率差距不断缩小。
表6 为不同废水初始pH 下废水COD 降解过程拟一级动力学模型参数。从表6可以看出,当pH0为3、6、9 时,其模型决定系数R2分别为0.998、0.999 和0.995,具有良好的拟合度。而当pH0为12 时,废水COD 降解的拟一级动力学模型R2为0.954,模型拟合度较差。当pH0为12时,废水pH随时间变化幅度较大,反应过程中氧化活性物质的组成及氧化能力都会有较大的变化,对氧化反应的动力学产生了较大的影响,因此,不同时间段的COD 降低速率与COD浓度的关系并不一致。

表6 不同废水初始pH下废水COD降解的动力学参数R2Table 6 Kinetic parameters R2 of COD degradation in wastewater at different initial pH
综合以上的实验结果可知,由于废水pH在电化学氧化过程中是一个持续变化的量,废水的初始pH对该废水中有机物降解效果的影响随着反应的进行逐渐减小,但有机物降解效果仍然在弱酸性(pH为3或6)条件下的更好。由于原废水的pH约为6,可不对该废水的初始pH进行调整。
2.2 最佳条件下的废水处理效果
在最佳反应条件下处理前后的模拟废水对比如图6 所示。处理后模拟废水COD 去除率为92.3%,COD 含量为246 mg/L,电流效率为32.3%,能耗为36.5 kW·h/kg。静置4 h后,其上清液的COD值低至0 mg/L。说明电化学氧化过程中存在一定的电絮凝作用,可去除该模拟废水中6%~7%的COD。通过稀释倍数法分析,废水色度由处理前的300 倍降为无色,色度去除率达100%。

图6 电化学氧化前(左)、后(右)废水图Fig.6 Wastewater before(left) and after(right)electrochemical oxidation
2.3 电化学氧化过程中有机物降解途径的探究
目前认为的电化学氧化途径包括两种:1)有机物在电极表面直接发生电子转移的直接氧化过程;2)以H2O、Cl-、SO42-等在阳极失电子生成的强氧化性活性物质(·OH、HClO、ClO-、·SO4-等)作为氧化媒介,与有机物发生氧化反应的间接氧化过程。
2.3.1 活性氯间接氧化作用
模拟的氯化钠有机废水主要由NaCl 和各种有机物组成,为了探究电化学氧化有机物过程中是否存在活性氯的间接氧化作用及其作用占比,使用除氯后的废水在电流密度为600 A/m2、反应温度为35 ℃、初始pH 为6 的条件下进行电化学氧化实验。图7为含氯和不含氯废水电化学氧化过程中COD去除率随时间的变化曲线。从图7 可以看出,在不含氯离子的废水中的COD 的去除率始终低于含氯废水,而且不含氯离子的废水在电解4 h 后COD 的降解速率明显放缓,其COD的去除率与含氯废水之间的差值进一步拉大,电解8 h 后不含氯废水COD 去除率由原含氯废水的92.3%下降为58%。

图7 含氯和不含氯废水电化学氧化过程中COD去除率随时间的变化情况Fig.7 Change of COD removal rate with time during electrochemical oxidation of chlorine-containing and non-chlorine wastewater
实验证明了氯离子的存在为废水中有机物的间接氧化提供了氧化活性物质,有效提高了废水有机物的降解效果,该模拟废水中约34.3%的有机物通过活性氯对有机物的间接氧化途径被降解。
2.3.2 自由基的间接氧化作用
在电化学氧化过程中,往往存在H2O 在阳极氧化形成物理或化学吸附态羟基自由基(·OH)的行为。·OH(E0=2.8 V)具有极强的氧化能力,可与大多数有机污染物发生快速的链式反应,将其氧化分解成CO2、H2O 和无机物。当电极为惰性电极(BDD、PbO2)时,所产生的·OH 更倾向于物理吸附于阳极表面,这种物理吸附的·OH 更易与有机物接触发生氧化反应。
物理吸附的羟基自由基[MOx(·OH)]主要由水的氧化[式(7)]形成,羟基自由基氧化有机污染物的反应,如式8所示[29]。
为了进一步探究是否有羟基自由基参与有机物的间接氧化过程,研究设计了羟基自由基掩蔽实验,通过在除氯后的废水中添加自由基掩蔽剂后电解,来探究掩蔽羟基自由基是否会对废水有机物的降解产生影响。实验以Na2CO3作为羟基自由基掩蔽剂[37],向除氯后的废水中加入0.1 mol/L的Na2CO3后进行电化学氧化实验,以除氯废水的电化学氧化实验为对照。图8为添加自由基掩蔽剂前后废水COD去除率随电解时间变化的曲线。由图8 可以看出,在添加0.1 mol/L的Na2CO3作为掩蔽剂后,废水COD去除率明显低于未添加掩蔽剂的实验组,电解8 h后废水COD 去除率由未加Na2CO3前的58%下降至34.6%。由此可知,在电化学氧化该高盐有机废水过程中,存在H2O 在阳极氧化生成的·OH 对有机物的间接氧化作用。

图8 Na2CO3对废水COD去除率的影响Fig.8 Effect of Na2CO3 on removal rate of COD in wastewater
综上所述,在Ti/PbO2阳极电化学氧化降解模拟废水有机物的过程中除了存在有机物在电极上的直接氧化作用外,还存在以活性氯和·OH 为主要氧化活性物的间接氧化作用,其中以间接氧化作用为主导。
2.4 一膜两室电解工艺
2.4.1 直接电解
NaOH 为一膜两室电解工艺的阴极产物。电解过程中阳极废水中的Cl-和有机物因无法透过阳离子交换膜被留在阳极室,而钠离子则在电场力的作用下透过阳离子交换膜去往阴极和阴极电解产生的氢氧根结合生成氢氧化钠。表7 为阴极室NaOH 浓度随电解时间的变化情况。从表7 可以看出,阴极NaOH 浓度随时间的增加而稳定提高,电解8 h 后NaOH 浓度从初始的1.24 mol/L 增加到2.21 mol/L,共产生10.5 g NaOH。

表7 阴极NaOH浓度随时间的变化情况Table 7 Change of NaOH concentration with time
留在阳极室的有机物则在阳极被氧化降解。图9为一膜两室电解法阳极废水COD去除率随电解时间的变化情况,并对比了2.2节最佳条件下的单室电化学氧化法。从图9 可以看出,在相同的实验条件下,一膜两室电解法对该氯化钠有机废水中有机物的降解效果明显低于单室电化学氧化法,电解8 h后COD去除率仅为51.2%。

图9 不同处理方法下废水COD去除率随电解时间的变化情况Fig.9 Change of COD removal rate with time at different treatment methods
图10 为一膜两室电解法阳极废水和单室电化学氧化法废水pH随电解时间的变化曲线。从图10可以看出,一膜两室电解法阳极废水pH与单室电化学氧化法废水的pH有较大差距,单室电化学氧化过程废水pH 随着反应的进行缓慢升高至8.3,整个反应体系pH维持在弱酸性至弱碱性范围内,而一膜两室电解法阳极废水pH 在反应开始后迅速降低并维持在3左右。

图10 单、双室电解过程废水pH随时间的变化情况Fig.10 Change of pH of wastewater in single and double chamber electrolysis processes with time
一膜两室电解过程中阳极液pH 维持在3 左右主要是因为反应过程中,Cl-在阳极被氧化生成的Cl2与水发生歧化反应后产生大量的氢离子使阳极液pH迅速下降。当Cl2在废水中溶解饱和后,废水pH维持稳定。
Cl2在溶液中主要以Cl(2aq)、HClO、ClO-和Cl3-形式存在,其存在形式随溶液pH 的变化如图11 所示[38]。从图11可以看出,当溶液pH大于8时,溶液中活性氯的主要存在形式为ClO-;pH 为3~8 时,溶液中活性氯的主要存在形式为HClO;当溶液pH 小于3时,溶液中活性氯的主要存在形式为Cl2。不同存在形式的活性氯氧化能力大小依次为:E0=1.50 V(HClO)>E0=1.36 V(Cl2)>E0=0.89 V(ClO-)[39]。因此,pH在3~8的活性氯氧化能力更强。

图11 不同pH下活性氯的存在形式[38]Fig.11 Existing forms of active chlorine at different pH
实验废水中氯化钠质量浓度为255 g/L左右,根据文献报道,Cl2在233~291 g/L 的氯化钠溶液中的溶解度为0.036 mol/L[40]。Cl2饱和溶解于300 mL 阳极液中的理论电解时间为29.43 min。即反应30 min后阳极液中Cl2溶解饱和,阳极液pH维持稳定,后续反应时间内生成的Cl2部分以气体形式溢出电化学氧化体系。而提高溶液pH可以促进Cl2与水的歧化反应,生成HClO 和NaClO 等物质,提高活性氯的含量。
综上所述,结合实验结果可知,在pH 为3~8 时提高溶液pH,不仅可以获得强氧化能力的活性氯,还可以提高溶液中活性氯含量,有利于有机物的降解。
2.4.2 加料电解
为了提高阳极COD去除率,根据2.4.1节的实验结果以添加NaOH 的方式提高一膜两室的阳极液pH。每隔0.5 h向阳极室添加0.5 g的NaOH,其他操作条件不变。
表8为间歇添加NaOH电解过程阳极液pH随电解时间的变化。从表8可以看出,间歇添加NaOH可以使阳极液pH 维持在4~6,较直接电解时的pH 维持在3左右有明显提升。
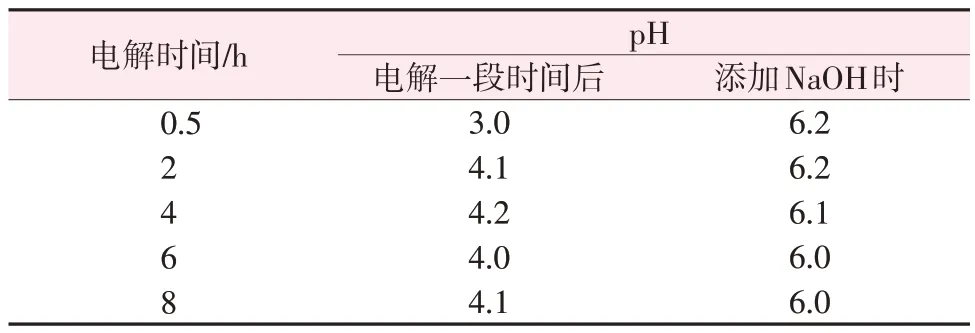
表8 阳极液pH变化情况Table 8 Change of pH of anolyte
图12为阳极间歇添加NaOH电解与直接电解过程阳极废水COD 去除率随时间的变化情况。从图12 可以看出,阳极液添加NaOH 后,废水COD 去除率明显高于直接电解,电解8 h 后废水COD 去除率较直接电解法提高了27.7%,可达到85.7%。这说明通过在阳极添加NaOH 使阳极液pH 维持在较高水平的方法可以有效提高废水有机物的降解效率。

图12 添加NaOH前/后阳极废水COD去除率随电解时间的变化Fig.12 Change of COD removal rate with electrolysis time before/after adding NaOH
间歇性向阳极室添加NaOH的操作方式在实际工业生产中难以应用,因此考虑通过管路向阳极室中持续添加NaOH溶液来替代NaOH的间歇加料。
将8 g NaOH 溶解在60 mL 的去离子水中,使用蠕动泵以7.5 mL/h的速率将该溶液持续加入阳极液中,其他条件不变电解8 h。图13 为不同加料方式下阳极废水COD 去除率随电解时间的变化情况。从图13 可以看出,采用连续添加NaOH 的操作方式在阳极液COD 降解效果上可达到与间歇式操作相近的水平,且有机物降解效果更加稳定,电解8 h后废水COD 去除率可达到87.2%,相较于间歇式添加NaOH 的85.7%也有所提高。因此可以证明在阳极液中连续添加NaOH的操作方式具有可行性。此时阴极共生成氢氧化钠11.33 g,电流效率为81.74%,电耗为4 kW·h/kg。

图13 不同NaOH添加方式下阳极废水COD去除率随反应时间的变化情况Fig.13 Change of COD removal rate of anode wastewater with reaction time at different NaOH addition methods
对一膜两室工艺生产的NaOH 进行杂质检测。表9 为不同反应时间下阴极NaOH 溶液的COD 值。从表9 可以看出,电解过程中有少量有机物进入阴极,随着电解的进行阴极NaOH 溶液COD 缓慢增加但始终维持在较低水平,8 h后的COD值为26 mg/L。
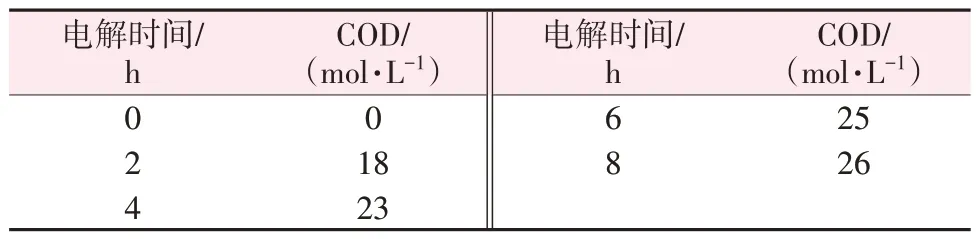
表9 不同反应时间下阴极液COD值Table 9 COD value of cathode solution at reaction times
阳极有机物在电化学氧化降解过程中会生成小分子有机酸从而更容易在浓度差的作用下穿过离子交换膜进入阴极液[41]。表10 为电解8 h 后阴极NaOH 溶液无机杂质含量。从表10 可以看出,阴极液中存在微量因电场力和浓度差而透过阳离子交换膜的杂质离子。因此,该NaOH 产品可应用于对钙镁离子和有机物含量无严格要求的工业生产。

表10 阴极液无机杂质含量Table 10 Contents of inorganic impurity in catholytem g/L
3 结论
1)通过单室电解研究了某香料企业氯化钠废盐中有机物的电化学氧化过程。研究表明,该废盐中有机物的电化学氧化降解过程符合拟一级动力学模型,电化学氧化降解该废盐中有机的最佳条件:电流密度为600 A/m2、反应温度为35 ℃、模拟废水初始pH 为6,在该条件下电解8 h,废水COD 去除率可达92.3%、色度去除率为100%、电流效率为32.3%。
2)除氯实验和羟基自由基掩蔽实验表明,该氯化钠废盐中有机物的降解,除了存在有机物在电极上的直接氧化作用外,还存在以活性氯和·OH 为氧化活性物的间接氧化作用,其中以间接氧化作用为主导。
3)单室电化学氧化和一膜两室电解实验结果对比研究表明,在含有大量氯离子的电化学氧化体系中,由于活性氯的存在形式和溶解度受体系pH 影响,维持体系pH在3~8更有利于提高有机物的降解效率。
4)通过电化学氧化法与膜法耦合,研究提出了一膜两室资源化处理氯化钠有机废盐工艺,不同于传统一膜两室电解法利用氯化钠废水制碱或降解废水中的有机物,该工艺实现了在阳极有效降解氯化钠废水中有机物的同时在阴极获取氢氧化钠。在单室电化学氧化的最佳条件下,电解8 h 阳极COD 去除率可达87.2%,阴极氢氧化钠总产量可达11.33 g。本研究可为氯化钠有机废盐的无害化处理提供参考。
5)由于膜性能及氯化钠模拟废水中有机物等杂质含量较高,一膜两室电解工艺阴极NaOH 产物含有少量有机物和杂质离子,NaOH 产物纯度可通过废盐预处理等方法以进一步提高。
猜你喜欢
中学生数理化(高中版.高考理化)(2020年3期)2020-05-30
山东冶金(2018年6期)2019-01-28
电子制作(2018年12期)2018-08-01
电镀与环保(2017年5期)2017-12-19
制造技术与机床(2017年12期)2017-02-02
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年2期)2017-01-20
电源技术(2015年9期)2015-06-05
