
肿瘤免疫标志物在乳腺癌免疫治疗中的作用机制及应用进展
2024-03-25 04:16杨凯曾天宇殷咏梅
药学进展 2024年2期
杨凯,曾天宇,殷咏梅
(南京医科大学第一附属医院/江苏省人民医院肿瘤科,江苏 南京 210029)
作为肿瘤药物治疗史上的第三次革命,免疫治疗被誉为目前最有希望的抗肿瘤手段之一。基于“免疫监视”理论,免疫治疗充分调动患者自身的抗肿瘤免疫系统,从而识别并清除“非己”肿瘤细胞[1];基于“肿瘤免疫编辑”理论,免疫治疗逆转“逃逸”、维持“均衡”甚至恢复“清除”阶段,从而长期有效控制肿瘤的进展[2]。近年来,免疫治疗已在黑色素瘤、肺癌和白血病等肿瘤中取得了显著的临床疗效,而传统被认为“弱免疫原性”的乳腺癌(breast cancer,BC),也在以免疫检查点(immune checkpoint,IC)为代表的三阴性乳腺癌(triple-negative breast cancer,TNBC)相关临床研究中取得了突破性进展。此外,免疫检查点抑制剂(immune checkpoint inhibitor,ICI)单药或联合化疗的新辅助治疗也有助于改善BC患者的预后[3]。然而,目前BC的免疫治疗仍面临诸多挑战,包括筛选敏感人群、提高疗效、预防耐药和免疫相关不良反应(immune-related adverse event,IRAE)以及寻找合适的药物靶点等[4]。
肿瘤免疫标志物是指可被客观衡量从而评估肿瘤免疫的生物学特征或指标,可用来预测BC免疫治疗的疗效从而筛选获益人群,监测免疫反应的过程从而预防IRAE,以及为BC免疫药物的研究提供靶点从而增强免疫治疗的疗效[5]。基于肿瘤免疫标志物的治疗,打破了既往按照不同瘤种进行统一规范化治疗的理念,而是根据标志物的表达情况进行精准化、个体化治疗。
肿瘤免疫标志物的表现形式多种多样,广义上参与肿瘤免疫各个环节的分子(如受体或细胞因子)、细胞(类型和比例)甚至微结构[如三级淋巴结构(tertiary lymphatic structure,TLS)]等均可作为潜在的标志物[6]。除了病理或生理产生的标志物外,还可人为诱导肿瘤免疫标志物的产生。例如基于BC瘤内菌的发现,基因工程细菌或病毒在特异性感染肿瘤的同时能表达相应的抗原作为免疫细胞识别肿瘤的标志,从而诱导免疫原性细胞死亡(immunogenic cell death,ICD)甚至编码产生ICI类似物[7]。此外,标志物的获取及检测手段、药物结构设计和给药方式等也在很大程度上影响了肿瘤免疫标志物在BC中的临床应用。本文阐述了肿瘤免疫关键的起止环节(即免疫识别和杀伤)中标志物的作用机制,包括肿瘤抗原的产生和释放、固有免疫和适应性免疫的效应,并基于作用机制总结了肿瘤免疫标志物的临床应用和前沿研究。
1 免疫细胞识别肿瘤抗原
1.1 肿瘤抗原的产生
肿瘤抗原是随着肿瘤发生发展而异常表达的肿瘤标志物,主要来源于基因转录水平的异常,也可来源于转录后RNA剪接失调或翻译后蛋白修饰紊乱[8]。当受到物理、化学或生物等致癌因素的影响时,肿瘤细胞会发生“经典遗传学”的基因突变,主要表现为原癌基因和抑癌基因的平衡被打破[9]。由于基因序列发生变化,肿瘤细胞可产生正常细胞不表达的抗原,即肿瘤特异性抗原(tumor specific antigen,TSA),其具有较高的免疫原性和个体异质性[10]。理论上突变的数量越多,即肿瘤突变负荷(tumor mutational burden,TMB)越高,产生的肿瘤抗原也越多,然而BC中高TMB不一定预示着理想的免疫疗效,这可能与肿瘤异质性有关[11]。
另一方面,肿瘤细胞会发生“表观遗传学”的基因表达异常,主要表现为相关基因表达的时空、数量和功能异常。由于没有发生基因序列的改变,肿瘤细胞会产生正常细胞表达的抗原,即肿瘤相关抗原(tumor associated antigen,TAA),例如用于BC分子分型的雌激素受体、孕激素受体和人类表皮生长因子受体2(human epidermal growth factor receptor 2,HER2),其具有较弱的免疫原性且诱导免疫耐受[12]。相比于TAA,TSA是BC中启动机体抗肿瘤免疫和T细胞效应器激活的关键[13]。然而,肿瘤抗原并非只是免疫系统识别肿瘤的“身份证”,某些肿瘤抗原在很大程度上促进了肿瘤的发生发展。例如,HER2阳性BC因高表达HER2分子从而加强生长因子信号介导的细胞增殖[14]。
1.2 肿瘤抗原的释放
在BC的肿瘤微环境(tumor microenvironment,TME)中,能量缺乏和代谢物堆积(如缺氧、葡萄糖缺乏、乳酸堆积和氧化应激)可诱导肿瘤细胞损伤(如内质网应激)和非凋亡调节性细胞死亡(尤其是ICD)[15]。肿瘤细胞损伤会表达损伤相关分子模式(damage-associated molecular pattern,DAMP)分子(例如内质网分子伴侣钙网蛋白和热休克蛋白),通过与模式识别受体(pattern recognition receptor,PRR)结合从而吸引并激活固有免疫细胞。肿瘤细胞死亡也会释放肿瘤内部抗原、三磷酸腺苷和高迁移率族蛋白1等免疫刺激因子从而促进免疫招募和浸润[16]。
然而,肿瘤细胞的死亡不一定都有助于BC的抗肿瘤免疫。研究发现,肿瘤细胞死亡释放的钾离子能抑制效应T细胞(effector T cell,Teff)的抗瘤作用[17],释放的前列腺素E2能抑制DAMPs对巨噬细胞和树突状细胞(dendritic cell,DC)的激活[18]。此外,肿瘤死亡也能诱导肿瘤细胞进入“警觉状态”。与固有免疫细胞类似,肿瘤细胞也能表达Toll样受体(toll-like receptor,TLR)和P2X4受体(P2嘌呤能受体的亚型)等PRRs,从而识别DAMPs分子并激活胞内核转录因子NF-κB和丝裂原活化蛋白激酶等信号通路,最终促进肿瘤细胞存活并产生炎症因子和趋化因子[19]。
2 免疫细胞杀伤肿瘤细胞
2.1 固有免疫的标志物
根据CD56表达的差异,可将自然杀伤(natural killer,NK)细胞分为以细胞毒作用为主的CD56dim和以免疫调节为主的CD56bright[20]。与T细胞类似,研究发现了调节性NK细胞、NK细胞耗竭以及组织驻留NK细胞等亚型。鉴于BC中NK细胞浸润的异质性,肿瘤中NK细胞的数量和类型可在一定程度上预测BC免疫治疗的疗效[21]。然而与Teff不同,NK细胞不表达特异性抗原识别受体,而是通过活化性杀伤细胞受体(activated killer cell receptor,AKR)和抑制性杀伤细胞受体(inhibitory killer cell receptor,IKR)信号的平衡来调节自身的活化状态(见图1A)。因此,NK细胞不需要抗原呈递,且具有更广谱、迅速和安全(可用于同种异体移植)的抗肿瘤作用,从而推动了NK工程细胞在BC中的应用[22]。

图1 免疫细胞表面抑制性和活化性免疫检查点及其配体Figure 1 Inhibitory and activating immune checkpoints and their ligands on the surface of immune cells
NK细胞主要通过AKR识别肿瘤表面的非主要组织相容性复合体(major histocompatibility complex,MHC)I类分子被激活,而通过IKR识别MHC I类分子被抑制[23]。为逃避Teff的识别,肿瘤细胞通常会下调MHC I类分子的表达,但MHC I类分子的表达缺失反而会介导NK细胞的杀伤,即“迷失自我”效应[24]。然而在BC的TME中,肿瘤浸润NK细胞的数量和功能会受到抑制(尤其是细胞毒性亚群)。例如,非经典HLA基因HLA-E和HLA-G被证实在BC中高表达,并能向NK细胞以及CD8+T细胞提供抑制信号,这可能与BC治疗中曲妥珠单抗耐药有关[21-25]。
BC中的巨噬细胞主要来源于循环单核细胞和组织驻留巨噬细胞,二者在TME的影响下形成肿瘤相关巨噬细胞(tumor-associated macrophage,TAM)[26]。在免疫反应早期,TAM主要发挥杀伤作用,高表达CD80、CD86、MHC II和CD68;而在免疫反应后期,TAM主要发挥免疫抑制作用,高表达CD206、CD204和CD163[27]。研究表明,TAM细胞频率与BC的免疫细胞浸润和免疫治疗反应有关[28]。与NK细胞类似,TAM通过“吃我”和“不吃我”信号的平衡来调节自身的活化状态(见图1B)。
2.2 适应性免疫的标志物
不同于急性感染的经典激活模式,BC中T细胞的激活分为2个阶段,即肿瘤引流淋巴结(tumor-draining lymph node,TDLN)中经典树突状细胞(conventional dendritic cell,cDC)抗原提呈的初始激活以及随后TME内的效应器激活。效应器激活主要包括3个信号:T细胞受体(T-cell receptor,TCR)-pMHC、共刺激分子和细胞因子[29](见图1C)。
目前,关于BC免疫的基础研究大多聚焦于CD8+T细胞和肿瘤组成性表达的MHC I类分子,然而CD4+T细胞的细胞毒作用和肿瘤诱导性表达的MHC II类分子也值得关注。研究发现,干扰素-γ(interferon-γ,IFN-γ)通过转录主调节因子Ⅱ型反式激活因子诱导肿瘤细胞表达MHCⅡ类分子,从而被CD4+细胞毒性T淋巴细胞(cytotoxic T lymphocyte,CTL)识别并杀伤[30]。与MHC I类分子不同,MHCⅡ类分子能结合多样性的抗原肽,从而促进CD4+CTL识别TSA[30]。更重要的是,研究发现MHCⅠ和MHCⅡ类分子在肿瘤免疫中受到独立调节,二者的表达可能对BC免疫治疗具有独立意义[31]。
在BC的肿瘤免疫中,肿瘤细胞可通过调节TME中IC的表达来逃避T细胞的杀伤,其中最重要的是上调负性共刺激分子[例如程序性死亡受体-1(programmed death receptor-1,PD-L1)],从而使T细胞维持在耗竭状态,即T细胞耗竭(T cell exhaustion,Tex)[32]。虽然Tex与T细胞失能在抑制性受体的表达上有重叠,但失能T细胞主要出现在免疫激活早期,而Tex则由晚期Teff转变而来[33]。有研究根据Ly108(由人转录因子TCF1表达)和CD69将Tex分为4个阶段,即耗竭前期1(TexProg1)、耗竭前期2(TexProg2)、耗竭中期(TexInt)和耗竭末期(TexTerm)。从TexInt开始,Tex逐渐失去TCF1的表达,即Ly108阴性,而抗PD-1治疗能恢复前3个阶段Tex的功能[34]。然而并非所有耗竭T细胞都失去功能,有研究发现部分T细胞耗竭后还能拥有多能性和再生性,即耗竭T细胞前体(Tpex)[35]。Tpex对维持Tex库和响应BC免疫治疗至关重要,进一步研究发现cDC1通过TCR信号使Tpex与持续存在的肿瘤抗原隔绝从而防止其进一步耗竭[36]。值得注意的是,之前研究认为肿瘤内Tex是响应BC免疫治疗的关键,然而后续研究表明响应BC免疫治疗的T细胞主要是外源性(TDLN)而并非局部TME中原本存在的[37]。最近有研究证实肿瘤细胞表达的PD-L1不能直接抑制T细胞毒性而是抑制T细胞浸润转移灶,DC和TAM等免疫细胞表达的PD-L1才是抑制T细胞毒性的关键,这部分解释了BC临床研究中PD-L1表达情况与免疫治疗疗效不匹配的问题[38]。
3 肿瘤免疫标志物在乳腺癌中的临床应用
除了寻找更合适的标志物外,如何最大程度发挥标志物在BC临床应用的价值更值得深思,包括标志物的获取及检测手段、药物结构设计和给药方式等。在标志物获取及检测手段方面,由于肿瘤的时间和空间异质性,来自活检等侵入性检查的标志物只能反映某一时刻部分肿瘤组织的静态情况,故其检查结果不具时效性且可能以偏概全;而来自血液等非侵入性检查的标志物,虽然便于实时动态反映肿瘤的整体情况,但由于表达水平低且干扰因素多,其结果难以检测且不够精准[39]。例如,对于BC的PD-1/PD-L1检测,除了传统活检外,BC的新型液体检测手段正在研究中,包括循环肿瘤PD-L1(PD-L1+CTCs)和外泌体型PD-L1检测[40]。在药物结构设计方面,BC中围绕肿瘤抗原的药物结构大多类似于抗体药物偶联物,即弹头[如靶向肿瘤抗原的抗体、TCR或嵌合抗原受体(chimeric antigen receptor,CAR)]加上弹药(如杀伤肿瘤细胞的毒素、药物或免疫细胞)以及二者的连接结构,其影响因素包括裂解性、脂溶性、药物抗体偶联比及旁观者效应等[41]。围绕IC的药物结构以大分子抗体为主,或可只保留Fab段来降低相对分子质量、增加肿瘤渗透率并降低免疫原性,但Fc段能诱导抗体依赖细胞介导的细胞毒性作用[42]。在给药方式上,鉴于肿瘤慢性病的特征和长期用药的需求,皮下瘤内注射(尤其对于较为浅表的BC)甚至口服给药是未来抗瘤药物的发展方向[43]。
鉴于单药疗效不理想,BC的免疫治疗更注重联合策略,包括联合化疗、靶向治疗和局部放疗来逆转BC的“冷肿瘤”状态。例如,ICD诱导剂联合ICI在增强免疫细胞浸润的基础上又能维持其抗瘤功能[44];ICI联合转化生长因子-β(transforming growth factor-β,TGF-β)抗体有助于药物渗透到肿瘤内部[45];双特异性抗体(bispecific antibodie,BsAb)同时靶向免疫细胞和肿瘤细胞的标志物,从而辅助免疫细胞接近并杀伤肿瘤细胞。此外,BsAb对于工程化免疫细胞的重定向作用能够逆转细胞免疫疗法的脱靶和耐药[46]。
3.1 基于肿瘤抗原的治疗策略
靶向肿瘤抗原的治疗策略主要包括ICD诱导剂、肿瘤疫苗、过继细胞疗法(adoptive cell therapy,ACT)和抗肿瘤抗体(见图2)。与TAA相比,TSA具有更高的免疫原性和肿瘤特异性,因而不易诱导免疫耐受、自身免疫反应和耐药[47]。TSA已成为BC肿瘤抗原治疗策略中极具潜力的标志物。然而,由于TSA具有较强的异质性和个体差异,确定特定突变与TSA之间的联系较为困难。目前已鉴定出的TSA数量远少于TAA,且针对TSA的个体化治疗需要更高的时间和经济成本[48]。因此,需要积极寻找肿瘤患者中共享表达的TSA,例如KRASG12D2突变产生的TSA[49]。

图2 基于肿瘤抗原的乳腺癌免疫治疗策略Figure 2 Tumor antigen-based immunotherapy for breast cancer
对不同来源肿瘤抗原优先级研究显示,从临床效果、免疫原性、特异性、肿瘤相关性、表达水平和阳性率以及细胞表达分布等方面来看,肿瘤内部抗原(主要是TSA)比肿瘤表面抗原或可溶性分泌抗原(主要是TAA)更具临床应用价值[50]。然而,BC中大多数基于肿瘤抗原的治疗策略(例如CAR-T细胞疗法和抗肿瘤抗体)很难直接靶向肿瘤细胞内部。因此,诱导肿瘤细胞死亡有助于肿瘤内部抗原的暴露[48]。此外,鉴于MHC能够结合胞内抗原并形成pMHC表达在细胞表面,TCR-T细胞疗法通过TCR-pMHC的结合能够识别大范围、低水平变异的肿瘤内部抗原。然而MHC表达的适应性调节及个体化,阻碍了TCR-T细胞疗法开发出更具临床价值的通用型TCR-T,这在一定程度上也限制了TCR-T的使用[51]。表1从作用机制的角度对肿瘤疫苗、ACT和抗肿瘤抗体进行了分类,对比了各种疗法的优缺点并重点总结了未来发展方向及相关临床研究[47-51]。
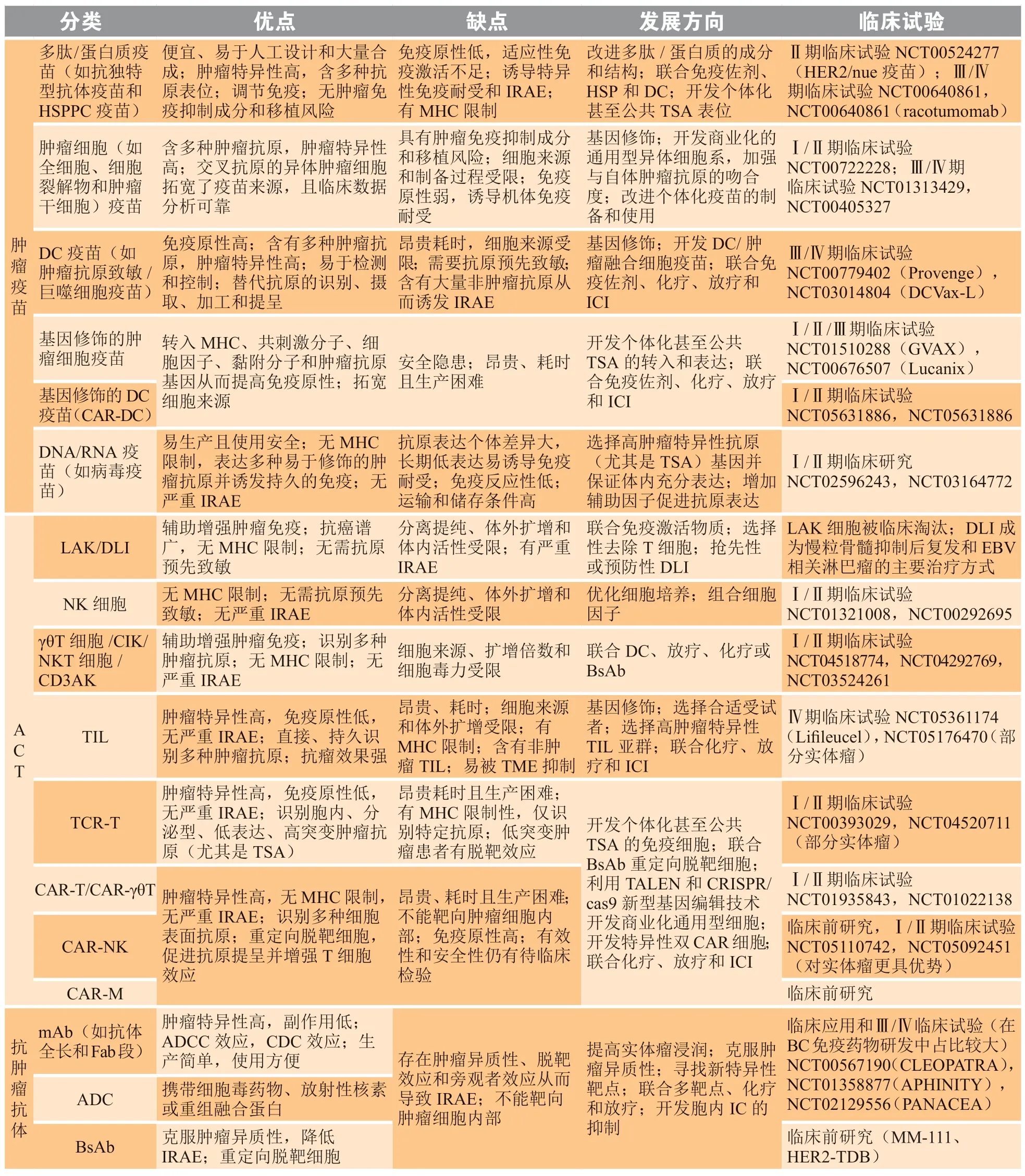
表1 基于肿瘤抗原的乳腺癌免疫治疗策略Table 1 Tumor antigen-based immunotherapy for breast cancer
3.2 基于免疫检查点的治疗策略
调节免疫细胞活化状态的IC主要包括NK细胞的AKR和IKR、巨噬细胞的“吃我”和“不吃我”受体以及T细胞的正性和负性共刺激分子[52-53]。表2总结了NK细胞、巨噬细胞和T细胞表面IC的受体-配体结合和分布情况,各个IC的作用特点及其在BC免疫治疗中的研究进展[52-53]。
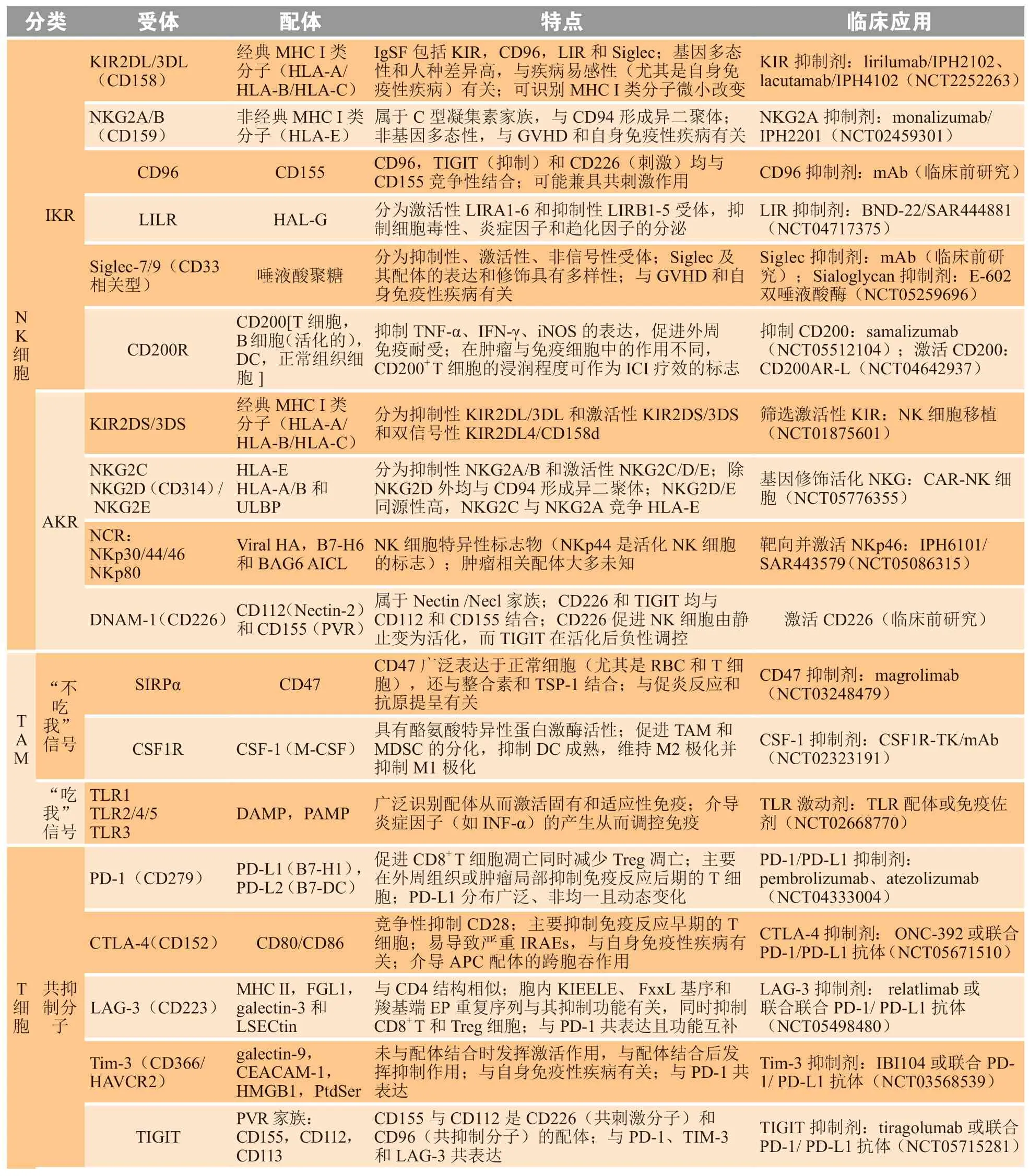
表2 基于免疫检查点的乳腺癌免疫治疗策略Table 2 Immune checkpoint-based therapeutic strategies against breast cancer
作为机体正常免疫调节器,IC对维持正常免疫耐受以及控制免疫反应的持续时间和范围至关重要,然而IC可被肿瘤“劫持”并异常表达从而介导免疫抑制。在BC免疫治疗中,为了筛选敏感人群、预防耐药以及副反应,在以关键IC为主要治疗靶点的基础上,许多研究报道了其他标志物用于辅助预测并监督肿瘤免疫反应,包括基因水平(如微卫星不稳定和TMB)、分子水平(如趋化因子和代谢相关分子)、细胞水平(如免疫细胞频率、TLS和TDLN)以及微生物水平(如病毒和肠道微生物)[54-55]。
ICI的抗瘤效果往往伴随着IRAE的发生,某些IRAE的出现可能预示着良好的免疫治疗效果[56]。然而IRAE的严重程度取决于共享IC表达的细胞类型、分化阶段和作用机制等。例如红细胞同样能表达巨噬细胞“不吃我”受体CD47,从而针对该靶点的BC免疫治疗易导致贫血[57]。此外,BC免疫治疗中抗CTLA-4主要影响了初始T细胞(Th0)从而过度激活免疫反应,而抗PD-1主要影响了晚期Teff,从而促使免疫反应恢复正常[58]。值得注意的是,有些IC的作用机制较为复杂,甚至对肿瘤具有双重作用,这导致单纯的阻断或加强活化信号不能取得理想的抗瘤效果。例如,随着BC的进展,肿瘤细胞自身可耐受TGF-β的作用,但免疫细胞仍受其抑制从而导致肿瘤的免疫逃逸,提示恢复BC对TGF-β的敏感性可能更有助于解决TGF-β抗体治疗所面临的问题[59]。
4 结语与展望
不同于以BC分子分型作为治疗指导,免疫治疗主要根据肿瘤免疫标志物的表达情况去调控肿瘤免疫反应的平衡。免疫治疗不再只是用于晚期BC患者的“没有办法的办法”,而是逐渐成为BC重要的治疗方式之一,积极用于辅助甚至新辅助治疗,并联合其他抗瘤药物[60]。目前,BC免疫治疗主要针对缺乏有效内分泌治疗和靶向治疗的TNBC,并在肿瘤疫苗、肿瘤抗体和免疫调节剂等方面取得了显著进展,但其大多数仍处于临床前或临床试验阶段,仅部分ICI被批准用于临床[61]。此外,BC免疫治疗仍存在诸多问题,包括筛选敏感人群、提高疗效、预防耐药和IRAE以及寻找合适的药物靶点等。这需要深入研究肿瘤免疫发生发展的各个环节(尤其是免疫激活和免疫效应),进而探索更加合适的肿瘤免疫标志物作为监测和治疗的靶点,同时进一步改进标志物的获取及检测手段、药物结构设计和给药方式等,从而最大程度发挥肿瘤免疫标志物的临床应用价值[4]。
猜你喜欢
中国生殖健康(2020年7期)2020-12-10
海南医学(2016年8期)2016-06-08
中华老年多器官疾病杂志(2016年9期)2016-04-28
西南医科大学学报(2015年1期)2015-08-22
医学研究杂志(2015年6期)2015-07-01
医学研究杂志(2015年7期)2015-06-22
癌变·畸变·突变(2015年3期)2015-02-27
现代检验医学杂志(2015年6期)2015-02-06
现代检验医学杂志(2015年5期)2015-02-06
郑州大学学报(理学版)(2014年2期)2014-03-01
