
酶催化杂Diels-Alder反应
2024-03-22 06:45:10王翠珍陈窕王健博
合成生物学 2024年1期
王翠珍,陈窕,王健博,2
(1 湖南师范大学化学化工学院,化学生物学及中药分析教育部重点实验室,植化单体开发与利用湖南省重点实验室,湖南 长沙 410081; 2 浙江大学基础医学院药物生物技术研究所,浙江大学医学院附属第二医院综合ICU科室,浙江 杭州 310030)
[4+2]Diels-Alder(DA)环加成反应是合成复杂天然产物的最有效的合成策略之一,而神奇之处在于,许多复杂结构的天然产物生物合成也包含了这一策略的应用[1-2]。DA反应是通过具有共轭结构的双烯体和提供不饱和键的亲双烯体发生反应,从而生产六元环己烯。该反应一次可以产生两个碳碳键,且在烯烃末端均有取代时,会新建四个相邻的手性中心,是一种构筑C—C键的有效方法,因此被广泛应用于复杂多环天然产物的全合成中[3-4]。DA反应自1928年Otto Diels和Kurt Alder发现以来,在近百年的时间里不断发展,现已成为有机合成的有力工具,且依旧是化学家关注的热点反应[5-6]。有关催化DA反应的报道不胜枚举,主要催化剂包括Lewis酸[7]、络合催化剂[8]、有机催化剂[9]等非酶催化剂,而在生物催化领域,这一类催化报道还相对较少,早期的催化剂主要包括抗体[10]和RNA[11]。天然酶催化DA反应的报道始于2011年[12],随后陆续也有其他新型DA酶被发现,多数酶类存在于天然产物的生物合成途径中,有关人工设计催化DA反应的酶也有报道。生物催化所具有的条件温和以及高选择性的优点,迅速引起大家对这个领域探索的兴趣[13-16]。
杂原子参与的DA反应称为Hetero-Diels-Alder(HDA)反应,是由杂双烯体和亲双烯体,或者是双烯体和杂亲双烯体进行DA反应得到六元杂环的反应[17]。按杂原子的不同,HDA反应可以分为氧杂[18-19]、氮杂[20-21]、硫杂[22]和磷杂[23-24]DA反应等,其中最常见的是氧杂DA反应和氮杂DA反应[25]。与经典DA反应相比,已知HDA加合物的生物催化方法报道相对较少[26]。HDA反应在合成各种杂环天然产物中发挥了重要作用,因此也成为构建复杂杂环化合物的有力工具。其中利用HDA合成天然活性产物的经典合成例子包括Leporin B(1)[27]、Leporin C(2)[28]等含有吡喃环的氧杂HDA产物(图1),以及(+)-Malbrancheamide(17)、(+)-Premalbrancheamide(18)[29]等含有双环[2.2.2]重氮辛烷的氮杂DA产物的合成(图2)。本综述主要对已报道的酶催化氧杂DA反应和氮杂DA反应进行归类整理,概述了HDA反应的研究进展,希望通过对合成途径以及催化机理的总结分析,促进该类生物催化剂的发展与应用。
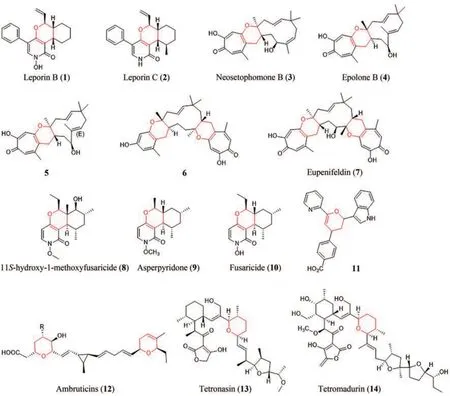
图1 吡喃类化合物Fig. 1 Pyran compounds

图2 含有双环[2.2.2]重氮辛烷的产物Fig. 2 Products containing bicyclo[2.2.2]diazaoctane rings
1 Diels-Alder反应的分类
DA反应可分为正电子需求反应和逆电子需求反应,双烯体通常是富电子的,而亲双烯体通常是缺电子的,它们之间的反应称为正常的正电子需求DA反应。当双烯体缺电子而亲双烯体富电子的时候,就会发生逆电子需求DA(IEDDA)反应[30-31]。可以利用前线轨道理论(FMO)来理解DA反应,其快速反应动力学主要由于双烯体的最高占有分子轨道(HOMO)和亲双烯体的最低未占有分子轨道(LUMO)之间的低能隙[32]。正常的[4+2]环加成被HOMOdiene-LUMOdienophile之间的相互作用所限制,由于这种限制的存在,需要催化剂的参与,以便有利于反应的发生,这通常是通过双烯体的亲核性或亲双烯体的亲电性的增加以加强HOMOdiene-LUMOdienophile之间的相互作用来实现,双烯体上的给电子基团可以提高HOMOdiene的能量,从而改善与亲双烯体分子LUMOdienophile的轨道相互作用。同样地,吸电子取代基的作用是降低亲双烯体LUMOdienophile的能量,从而增加与双烯体HOMOdiene的轨道混合,促进环加成反应。而逆电子需求则相反,是被HOMOdienophile-LUMOdiene的相互作用所限制(图3)[33-35]。对于化学合成来说,通常会用到Brønsted酸[36]、Lewis酸[37]、手性磷酸[38]等催化剂,来降低LUMO或者是提高HOMO,从而增加HOMO-LUMO的相互作用,达到提高活性的效果[39-40]。而酶催化DA反应生物合成中,除了未被验证的酶外,已被证实的催化DA反应的酶大多是正电子需求,如PyrE3[41]、CghA[42]和MycB[43]等,而IEDDA反应则报道较少,IccD3[44]、MalE[45]已被证实是催化发生IEDDA反应的酶。

图3 Diels-Alder反应的分类Fig. 3 Classification of Diels-Alder reactions
2 氧杂Diels-Alder反应
吡喃类化合物是指含有一个氧原子的六元杂环化合物,广泛存在于真菌以及植物中,具有多种抗癌活性,因此该类化合物也成为人们关注的焦点。其中,二氢吡喃环和四氢吡喃环是天然产物中普遍存在的结构特征。从生源途径分析,HDA反应被认为是构建吡喃环的关键生物合成反应,也是目前制备这一类化合物的主流方法[46-47]。
2.1 二氢吡喃类化合物的生物合成
二氢吡喃类化合物包括具有细胞毒和抗真菌特性的Leporin B(1)[48]、具有抗肿瘤活性的Neosetophomone B(3)和Eupenifeldin(7)[49]、基质溶素抑制剂和潜在的抗关节炎药物Epolone B(4)[50]、潜在的降糖药物Asperpyridone A(9)[51]等(图1)。基于其生物活性及结构特征,该类化合物生物合成研究的热度更是不断上升,取得了不少令人瞩目的研究结果。
2015年Jeffrey W. Cary等[52]对丝状真菌黄曲霉(Aspergillus flavus)的基因组进行了研究,发现lep基因簇导致了Leporin B(1)以及去羟基前体Leporin C(2)的生物合成,但并未对其合成途径进行深入研究。
2017年,唐奕教授课题组[27]在此基础上,将已报道的黄曲霉中Leporin B(1)生物合成基因簇,转入构巢曲霉(Aspergillus nidulans)中异源表达,以实现对Leporin B(1)合成途径的解析。研究发现底物苯丙氨酸(21)在聚酮合酶-非核糖体肽合酶(PKS-NRPS)LepA、烯酰还原酶(ER)LepG和用于环扩展的P450酶LepH的共同作用下生成22[53],然后在短链脱氢酶(SDR)LepF作用下进一步生成23。而23在无酶情况下,自发反应得到分子内DA以及HDA反应的混合物,反应无立体选择性。据此推测,反应立体选择的控制必然来源于酶促,而其中最为可能的候选者为LepI,其归属于SAM(S-腺苷甲硫氨酸)依赖的O-甲基转移酶家族。随后经实验确认,当反应体系中存在LepI时,只有Leporin B(1)的前体Leporin C(2)生成,进一步加入P450 LepD得到了预期的最终产物Leporin B(1)。为进一步分析LepI,作者分离出23的两个异构体,结果发现在无酶对照中依旧会自发反应,这与前面实验结果保持一致。在添加LepI后,23在辅助因子不存在的情况下完全转化为2,由此证明LepI是23转化成2的关键因素,具体反应过程需要先立体选择性脱水生成(E)-24,然后再经HDA反应生成2(图4)。同时还发现该反应伴随有分子内Diels-Alder反应,证明LepI具有催化脱水、分子内Diels-Alder反应、HDA反应以及反Claisen重排的多种功能。为了进一步解析LepI催化机理,作者进行了密度泛函理论(DFT)计算。计算结果发现与(Z)-24相比,(E)-24转化为2的自由能要低得多,这与实验结果相符。(E)-24经HDA反应和分子内DA反应分别转化为2和25的过渡态能量都较低(分别为-11.2 kcal/mol和-17.1 kcal/mol,1 kcal=4.2 kJ),这也是两种产物均能产生的原因。但是相对而言,经HDA反应的能垒更低,产物2占比更大[54-55]。

图4 Leporin B(1)的生物合成途径Fig. 4 Biosynthetic pathway of Leporin B (1)
随后,LepI的催化机理引发了人们的广泛兴趣,目前也有数例结构以及催化机理的研究报道[54,56-60],郭瑞庭教授课题组[56]通过与来自草酸青霉(Penicillium oxalicum)的一种SAM依赖的O-甲基转移酶OxaC结构比较,进一步解析了LepI的晶体结构。研究发现,与典型的Ⅰ类甲基转移酶一样,LepI也折叠成N端和C端结构域。程伟教授课题组[59]则发现α1与α2螺旋的缺失,会完全消除酶的活性,这也意味着LepI与其他同源O-甲基转移酶中观察到相同的二聚体组织不同,其二聚体会通过α1和α2螺旋相互作用形成四聚体,底物周围阳离子残基有增强反应活性的功能。此外,SAM与C端的Rossmann折叠结合,化合物2位于N端和C端界面上形成的一个大口袋中,靠近SAM结合位点,SAM的腺苷环和氨基酸分别通过π-π相互作用和氢键结合在口袋中,结合强度也对LepI的反应活性产生影响。同时SAM带正电的部分可能与22的苯基部分和吡啶环形成阳离子π-π相互作用,这也证明SAM对酶的催化功能至关重要。虽然对SAM的具体功能还未研究透彻,但为后续的实验提供了有力的参考与借鉴。
胡友财教授课题组[28]利用邻醌甲基托丙酮(29)与葎草烯的HDA反应合成Neosetophomone B(3)、Epolone B(4)和Eupenifeldin(7)。该课题组最初从微紫青霉(Penicillium janthinellum)中分离出3和4,证明微紫青霉中存在这一类化合物的生物合成途径。进一步对其基因组进行生物信息学分析,鉴定出一个NR-PKS基因簇,将其命名为eupf簇[61-62]。为验证eupf基因簇并阐明3的生物合成途径,将该基因簇转入构巢曲霉(Aspergillus nidulans)中进行异源表达。结果发现非还原聚酮合酶(NR-PKS)EupfA、FAD依赖的单加氧酶EupfB以及α-酮戊二酸依赖的双加氧酶EupfC共同催化下,生成了化合物29。将短链脱氢酶(SDR)EupfE于大肠杆菌(Escherichia coli)中异源表达,纯化后与29以及NADPH孵育,得到新产物30。而EupfG和细胞色素P450单加氧酶EupfD共表达后,发酵液中发现有33的产生,单独表达EupfG则会产生32。化合物32和33具有Z型烯烃键,这与3中烯烃的Z型键一致,以上结果表明,EupfG是一种新的倍半萜合酶。此外,该课题组利用EupfF的同源酶EupF[63](相似度81%/同源度67%),以及30为底物进行了体外实验,并用甘油来捕获31,确定了31的结构。实验发现EupF催化了30和33向3的转化,而化合物3是对映体纯化合物,这就表明EupF直接控制HDA反应的立体选择性。而后将EupfD在构巢曲霉中表达,34转化为非天然产物35,并检测到33自发与35发生HDA反应,生成Epolone B(4)及其异构体5(图5)。同时,研究发现EupfF与已知分子内DA酶不同,其并没有可识别的辅因子结合位点。最后通过密度泛函理论(DFT)计算发现,对应两条途径的过渡态能量都相对较低,加成的两分子处在距离较近的合适构型状态,因此环加成极易发生。遗憾的是,在30和33为底物实验中均未检测到双HDA加合物Eupenifeldin(7)。随后,Carsten Schotte等[64-65]报道了来自暗球腔菌属(Phaeosphaeriaceaesp.)的CF-150626产生了化合物6。通过对对应基因簇测序,将其命名为eup2,然后对eup2和来自乳产孢杆菌属(Leptobacilliumsp.)CF-236968的同源基因簇pyc进行了探究,成功利用PycR1合成了双HDA加合物36,该化合物进一步在米曲霉菌(Aspergillus oryzae)中转化成了产物6(图5)。虽然体外合成未能完成,且对7合成途径的探究尚存在欠缺,但仍成功实现了体内双HDA加合物的合成。
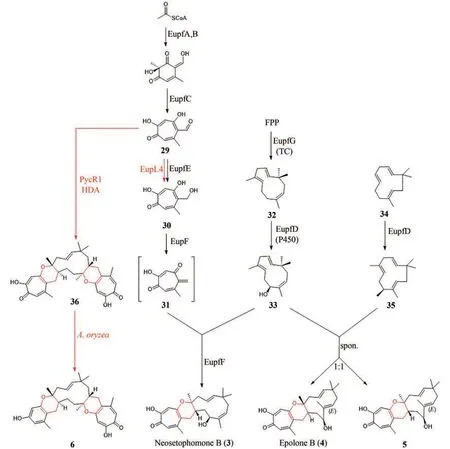
图5 Neosetophomone B(3)与Epolone B(4)的生物合成途径Fig. 5 Biosynthetic pathway of Neosetophomone B (3) and Epolone B (4)
2017年,曹树更教授课题组[66]利用真菌FT1061(Camporesia sambuci)和FT1062(Epicoccum sorghinum)共培育获得了新的N-甲氧基吡啶酮类化合物8,该产物为Fusaricide(10)的类似物(图1)[67]。2020年,唐奕教授课题组[68]在此基础上,报道了Fusaricide(10)的前体Asperpyridone A(9)的生物合成途径。作者通过基因组搜索,发现了含有聚酮合成酶-非核糖体肽合成酶(PKS-NRPS)的基因簇,以及催化HDA反应的EpiI。具体生物合成途径包括起始底物在PdxC(PKS-NRPS)和烯酰还原酶(ER)PdxD催化下反应生成37,然后进一步利用P450酶PdxA催化环扩展生成38。接下来在短链脱氢酶(SDR)PdxG和辅因子NADPH的存在下,38被还原为醇39。实验发现,在溶液中,39易发生非酶脱水生成(Z)-QM(40),而PdxG孵育过长会产生42。当加入O-甲基转移酶(OMT)PdxI或者其同源簇AdxI和ModxI时,主要发生Alder-ene反应形成43;当体系中加入EpiI或者其同源簇UpiI和HpiI时,周环发生选择性切换,经HDA反应生成41(图6)。这两种酶催化具有很强的周环选择性,不能相互转化。接下来作者又对PdxI和HpiI以及两种酶和底物与产物复合物的晶体结构进行了分析实验,发现EpiI为非SAM依赖的酶,且PdxI突变体(V413M)和EpiI双突变体(M411V/T231A)表现出相反的周环选择性[69]。

图6 Fusaricide(10)前体Asperpyridone A(9)的生物合成途径Fig. 6 Pathway for synthesizing the precursor Asperpyridone A (9) of Fusaricide (10)
从以上研究可知,催化HDA反应的天然酶相关基因簇,通常含有一种聚酮合酶(PKS)、一种短链脱氢酶/还原酶(SDR)、一种烯酰还原酶(ER)、两种P450(一种用于环扩展,一种用于羟基化)以及一种O-甲基转移酶(OMT)。
除天然酶外,也有通过人工设计酶实现HDA反应的报道,例如,Donald Hilvert教授课题组[70]利用人工金属酶HDA酶MID1sc制备二氢吡喃类化合物11。实验发现,利用44和45合成11的反应是一个竞争反应,可以通过竞争的分子间DA和HDA反应分别生成46和11(图7)。通过DFT计算表明,Zn(Ⅱ)(HO-)(H2O)2的加入使环加成反应从缓慢一致的反应转变为快速分布反应,并有利于内型HDA产物的生成。实验发现,模板MID1sc蛋白无法催化44和45的转化。通过DFT计算发现如果在位于含锌离子的结合袋两端增加两个突变E32L和K68W,可以在保持蛋白质稳定性的同时改善底物结合和过渡态稳定性,由此得到酶DA0,实验验证该突变体具有一定的目标活性。在对DA0的几个残基进行了四轮突变后,最后得到活性较高的十二突变体DA7。通过反应动力学测试发现,DA7催化效率相对于DA0提高了5个数量级以上,且具有高度的立体选择性,只生成内型HDA产物。随后,通过对DA7的晶体结构解析,对接计算和分子动力学(MD)模拟发现,只有产物11的内型异构体的过渡态适合结合口袋,同时与Zn(Ⅱ)配位强且没有发生空间冲突。最终利用该优势突变体的催化活性,成功实现了11的制备[70]。

图7 人工锌金属酶DA7催化HDA反应Fig. 7 HDA reaction catalyzed by the artificial zinc metalloenzyme DA7
2.2 四氢吡喃类化合物的合成
一些重要药物,如红霉素、双氢链霉素等分子以及许多碳水化合物都含有四氢吡喃环结构[71-72]。如来自黏菌多酮衍生的Ambruticins(12)(Ambruticin F和S等)家族,包含了两种杂环(图1),具有特殊的生理活性,其中Ambruticin S对多种真菌病原体表现出强大的抗真菌活性[73-74]。链霉菌属的Tetronasin(13)被用作抗生素和抗寄生虫剂[75],Tetromadurin(14)具有抗逆转录病毒和抗疟疾活性等作用[76],该类化合物重要的药用价值引发了人们极大的合成兴趣。
Peter F. Leadlay教授课题组通过测序以及生物信息学分析Tetronasin(13)生物基因簇发现,其中存在两种[4+2]环化酶Tsn11和Tsn15,与被证明是四毒霉素生物合成所必需的tmn同源[77]。为了验证两种酶的功能,作者首先在黄色长孢链霉菌(Streptomyces longisporoflavus)中敲除两种酶的编码基因后发现了代谢产物48的积累,证明48是最终产物的前体。随后以6-癸氨基-2-氟- 3-氧己酸甲酯(47)作为化学探针[78](图8),确定了PKS结合中间体的存在。接下来为了验证其体外活性,将两种酶表达纯化,并以48为底物进行体外实验,发现Tsn11催化HDA反应的发生,产生了氧化二烯中间体49,接着,Tsn15催化重排,形成四氢吡喃环,并分解氧化二烯部分,产生Tetronasin(13)(图9)。对Tsn11蛋白序列分析发现,Tsn11与生物合成途径中催化二烷基十氢化萘形成的VstK、PyrE3和ChlE3蛋白序列相似,而Tsn15则类似于类DA环化酶的第二家族VstJ、PyrI4和AbyU[79-82]。为进一步阐明催化机理,该研究团队对Tsn15的晶体结构进行了解析,发现反应过程中通过周环重排产生四氢吡喃环的机制,类似于其同源酶AOC2[83]和[4+2]环化酶PyrI4[82]和AbyU[84]。另外在Tsn15与底物共晶中,发现49羟基脱水生成50。由于Tsn15的活性位点更易与49形成氢键,因此49更有可能是Tsn15的底物。虽然确切机制尚不清楚,但为后续的进一步探究提供了有力的基础[77,85]。
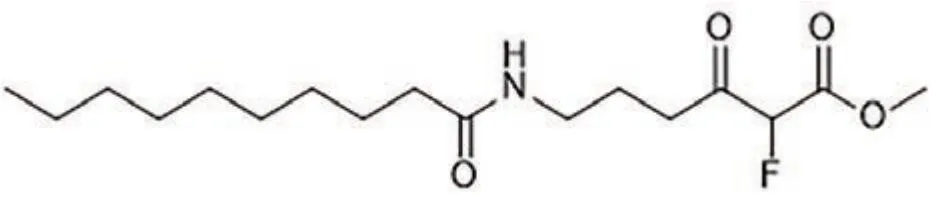
图8 6 -癸氨基-2-氟-3-氧己酸甲酯(47)Fig. 8 Methyl 6-decanamido-2-fluoro-3-oxohexanoate (47)
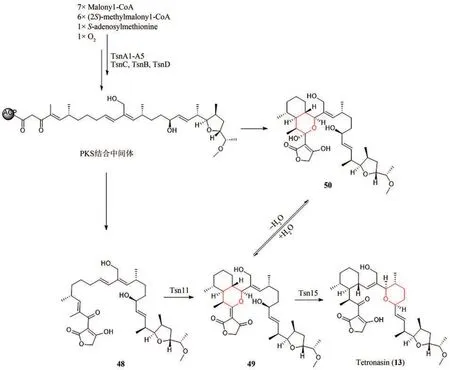
图9 Tetronasin(13)的生物合成途径Fig. 9 Biosynthetic pathway of Tetronasin (13)
2020年,该实验室又对与Tetronasin(13)结构高度相似的Tetromadurin(14)生物合成途径进行了报道[85-86],确定该化合物生物合成基因簇为mad,并确定与基因簇tmn和tsn同源[77],合成途径也类似,其中环化酶为Mad10和Mad31,可以实现前体51到54的转化,最后Mad30催化54进一步羟基化生成目标产物14(图10)。
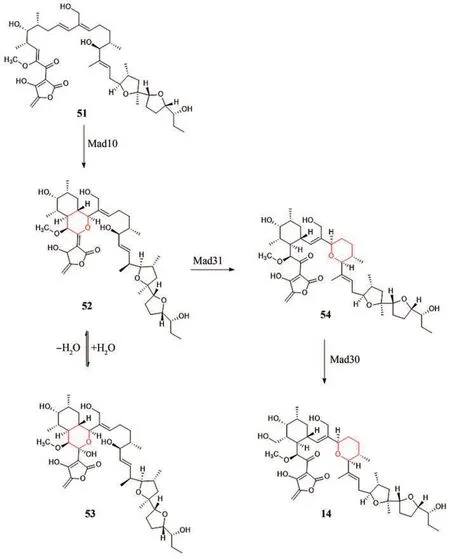
图10 Tetromadurin(14)的生物合成途径Fig. 10 Biosynthetic pathway of Tetromadurin (14)
综上可知,酶催化四氢吡喃类化合物的合成,一般包含两种环化酶,一种催化HDA反应,另一种则催化分子结构的重排,同时生物合成途径与合成机理都大同小异。通过对这些研究成果的分析,可以为人们后续的探索和研究提供借鉴。
3 氮杂Diels-Alder反应
来自于真菌曲霉属和青霉菌属中具有独特双环[2.2.2]重氮辛烷核心结构的吲哚类生物碱,因其有趣的化学结构和生物活性而受到越来越多的关注[87-89]。不同种类的吲哚生物碱具有不同生物学特性和药理活性,例如:具有抗癌作用的Stephacidin A(15)[90]、具有杀虫作用的Notoamide P(16)[91]、具有钙调素抑制作用的Malbrancheamides(17)等(图2)[92]。而这些天然产物的双环[2.2.2]重氮辛烷核心的生物合成途径一直以来广受关注。1970年,A.E.A.Porter和P.G.Sammes[93]首次提出该类结构来源可能通过分子内[4+2]DA反应产生,引发了人们对DA反应合成吲哚生物碱的关注。而目前所用手段大多数基于有机合成,相对而言,酶催化DA反应合成吲哚生物碱的报道甚少。
2016年,Sachiko Tsukamoto教授课题组[94]对多种吲哚生物碱的合成途径进行了汇总,并利用真菌培养分离出7种吲哚生物碱,提出了构建这些天然化合物可能的合理生物合成途径,但并未对其中酶的催化机理进一步研究。2019年,Robert M.Williams教授课题组[29]报道了Malbrancheamide(17)的生物合成途径(图11)。该课题组为了在化学上验证17的生物合成途径,首先通过有机合成得到了中间体61,进而经酶催化HDA反应,得到了17的前体(+)-Premalbrancheamide(18),由此表明61可能是合成最终产物的中间体。同时依据实验结果分析,提出了好氧和厌氧两种可能的生物合成途径。随后,该课题组进行了生物合成途径的体外重建,首先MalG催化L-脯氨酸和L-色氨酸偶联生成56,产物自发缩合脱水经57得到58。在有氧条件下,58自发氧化产生60,在异戊烯基转移酶MalE(来自mal基因簇)存在时,60经异戊烯基化产生两性离子61。随后氧化还原酶MalC先将61还原成62,进一步催化分子内DA反应,产生18,证明MalC具有还原和催化分子内DA反应的双重功能;在厌氧条件下,MalE催化58产生59,然后MalC先将59还原成62,接着催化发生分子内DA反应,产生18,18可以通过MalA催化进一步生成17(图11)。研究发现,与好氧条件相比,厌氧条件下该反应的效率相对较弱,这表明相对于58,61才是MalC的最适底物。同时发现NADPH为MalC的最适辅因子,使用NADPH的催化效率(kcat/Km)比利用NADH高10倍。为了进一步探究分子内DA酶MalC的催化机理,对其晶体结构进行了探究。但是在获取MalC的复合物晶体结构时没有成功,因此改为分析同源酶PhqE与底物61的复合物晶体结构,并进行分子动力学模拟和定点突变实验,由此提出了MalC的催化机制。MalC中关键残基Asp108与Arg130发生相互作用,稳定Arg130给出质子后的构象,Arg130作为质子供体与NADPH核糖的2′-OH结合促进该辅因子的结合。NADPH是电子供体,Asp165可以稳定61的正电荷,促进18的形成。IMDA反应的立体控制主要由形状互补驱动,底物与Trp168之间的接触,以及辅因子结合位置在IMDA过程中起着关键作用(图12)[95-96]。
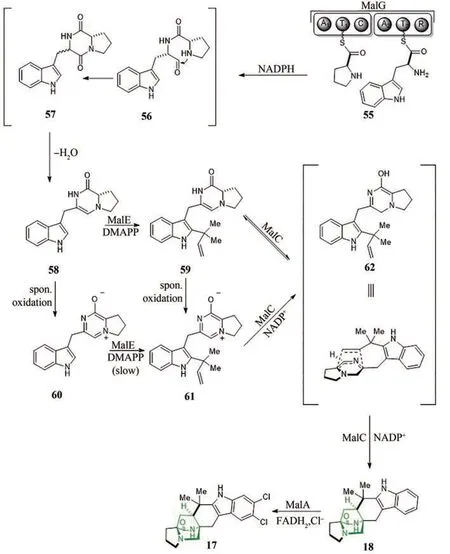
图11 吲哚生物碱Malbrancheamides(17)的生物合成途径Fig. 11 Biosynthetic pathway of indole alkaloid malbrancheamides (17)
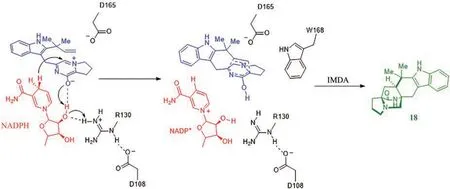
图12 MalC/Phq催化Diels-Alder反应的催化机理Fig. 12 Catalytic mechanism underlying the MalC/PhqE-catalysed Diels-Alder reaction
近期,K.N.Houk、David H. Sherman以及高雪教授课题组[45]发现NmrA类酶CtdP可以作为DA酶催化立体选择性合成(+)-Precitrinadin A(19)。为了验证其催化作用,将其表达纯化后,体外实验发现辅因子NADP+在CtdP催化过程中必不可少,且CtdP催化α-反式选择性IMDA环化。底物63在CtdP与辅因子NADPH作用下,生成α-反式中间体19,19可以进一步形成20。而在无酶条件下,63会自发生成非对映异构体64和65(图13)。对晶体结构分析发现,大体积残基Trp160、Phe170和Phe174发生了空间位冲突,表明这些残基可以在空间位上阻碍syn构象的形成。DFT计算表明CtdP催化的α-反式选择性IMDA环加成是通过氧化还原途径介导发生的,在该途径中,NADP+氧化降低了LUMOdiene,从而实现了IEDDA反应,这也证明CtdP为双功能氧化还原酶。通过MD模拟,NADPH会与63的氧化中间体形成氢键,阻止底物反转形成其他构象。据此提出了CtdP依赖于NADP+/NADPH的催化的逆电子需求DA反应的氧化还原机制(图14)。这是第一个报道的具有立体选择性催化HDA反应生成吲哚生物碱的双功能氧化还原酶[97-101]。

图13 吲哚生物碱(+)-Precitrinadin A(19)的生物合成途径Fig. 13 Biosynthetic pathway of indole alkaloid (+)-Precitrinadin A (19)

图14 CtdP催化的IEDDA反应的催化机理Fig. 14 Catalytic mechanism underlying the CtdP-catalysed IEDDA reaction
虽然酶催化合成吲哚生物碱双环[2.2.2]重氮辛烷核心结构的报道较少,但因为其合成价值,化学合成的方法有诸多研究。出于对绿色高效以及高选择性的制备方式需求,酶催化合成途径仍值得深入探索。
文中提到的HDA酶及其产物总结于表1。

表1 HDA酶及其产物Table 1 HDA enzymes and products
4 总结与展望
本综述主要对酶催化氧杂DA反应和氮杂DA反应的相关研究成果进行了概述,包括生物合成途径以及催化机理的分析。通过对已报道的HDA酶进行分析可以发现,对于二氢吡喃类化合物通常由一种聚酮合成酶(PKS)充当HDA酶的角色,催化HDA反应的发生,而四氢吡喃类化合物则由两种[4+2]环化酶协同作用催化产物的合成;而对于杂氮DA反应合成吲哚生物碱,辅因子NADPH的存在极其重要,与其他残基共同决定了产物的构象。
近年来,酶催化HDA反应获得了广泛的关注,得到了较快的发展。但不可否认的是,大多已报道的HDA酶的催化机理还没有被分析透彻,且多数HDA酶仍有待分离和表征。通过这些杂原子参与的酶催化DA反应合成杂环天然产物的结构分析发现,核心结构大同小异,所涉及的HDA酶具有较高的同源性,结构和功能以及催化机理极为相似,这也为后续HDA酶的进一步开发提供了参考。分析新的HDA酶的结构、功能以及催化机理等时,可以参考其同源酶,以此来降低分析难度。另外,酶催化HDA反应生物合成杂环天然产物的相关酶,大多数已经失去了功能的混杂性,只立体选择性催化[4+2]DA反应产物的生成,目前已知的唯一例外是CtdP,该酶属于氧化还原酶家族,且具有双功能[45]。
另外,除了氧杂DA反应和氮杂DA反应,关于其他杂原子,例如S或P等参与的DA反应大多以非酶催化剂催化反应的发生,而酶催化杂硫和杂磷DA反应的相关报道很少,已报道的HDA酶是否可以应用于催化含其他杂原子杂环的合成,这方面的研究还有待开发。
HDA反应作为合成天然杂环极其重要的工具,利用酶催化绿色、高效、高选择性地合成杂环天然产物已成为人们关注的重点,并希望能够对其合成途径、结构、机理进行深一步的挖掘。然而大多数HDA酶有着较高的底物特异性和立体选择性,且其底物谱非常狭窄,甚至一种HDA酶仅能实现一种杂环天然产物的合成,这就使得酶的应用受到了很大的限制,难以实现酶的使用价值最大化。因此,在之后的HDA酶研究中,一方面,研究者们可以通过定向进化等方法,对酶进行改造,还可以考虑对HDA酶同源重组进行基因融合,以实现已报道的HDA酶底物谱扩大化,甚至实现酶对不同类型底物的识别,从而得到更加多样、更加复杂的天然杂环产物,如已报道的PdxI[68]和DA7[70]等;另一方面,可以对已报道的HDA酶的同源酶进行深入挖掘,通过对比、实验以及MD或者是DFT等计算,实现对同源酶的性能、结构、催化机理以及产物合成途径等的探究;同时,要关注已报道过或者是新发现的能够产生HDA产物的生物,通过对该生物体内未验证其作用的基因进行测序,异源表达以及体外实验等,实现对新酶的探索。另外,基于天然酶的序列-结构-功能关系的解析,可以使用Rosetta等技术设计新的HDA酶,以获取功能更为强大的新酶。随着对HDA酶研究的不断进行,终将实现对HDA酶的结构、功能以及催化机理的深入了解,并指导天然以及非天然杂环产物的生物合成,成功实现合成的简单化和多样化。
猜你喜欢
新作文·中学作文教学研究(2022年4期)2022-08-25 02:38:48
化学工程师(2022年5期)2022-05-11 06:26:16
太原科技大学学报(2020年3期)2020-06-22 01:27:26
西北农林科技大学学报(自然科学版)(2019年8期)2019-07-17 02:43:32
合成化学(2015年9期)2016-01-17 08:57:14
烟草科技(2015年8期)2015-12-20 08:27:14
支点(2015年11期)2015-11-16 10:25:03
遗传(2015年5期)2015-02-04 03:06:55
海洋科学(2014年12期)2014-12-15 03:35:00
影像科学与光化学(2014年3期)2014-03-11 16:03:01
