
14例孤独症谱系障碍合并癫痫的临床表现及遗传学分析
2024-03-14 11:45:50朱书瑶江南静夏利聂晶樊军彭晓曾兰陈艾罗泽民王齐艳
中国生育健康杂志 2024年2期
朱书瑶 江南静 夏利 聂晶 樊军 彭晓 曾兰 陈艾 罗泽民 王齐艳
孤独症谱系障碍(autism spectrum disorder,ASD)和癫痫是儿童常见的神经系统疾病,估计全球发病率为0.5%~1%[1],经常共同存在。遗传学、神经影像学及神经病理学等多学科研究都提示遗传因素是这两种疾病的重要病因,ASD和癫痫之间的联系一直都无定论。本研究评估了14例ASD患儿合并癫痫的临床症状及遗传因素,以探讨两者之间的关系。
对象与方法
1.研究对象:选择2019年9月至2022年8月四川省妇幼保健院儿童保健科门诊诊断ASD,经神经内科确诊合并癫痫的14例患者作为研究对象进行病因分析。ASD患儿诊断标准:由主要抚养者填写孤独症儿童行为量表(ABC),ABC评分≥53分为筛查阳性,由儿保科医生采用儿童孤独症评定量表(CARS)进行评估,CARS得分≥30分的患者进一步由2名儿童发育行为专科医师逐一进行病史询问及行为观察,符合美国精神障碍诊断统计手册第5版(DSM-Ⅴ)诊断和分型者最终确定为ASD患儿。癫痫患儿诊断标准[2]:符合国际抗癫痫联盟(ILAE)2017年关于癫痫的诊断分类标准的癫痫患儿。排除标准:合并其他精神类疾病者,如精神分裂症、转换障碍等。本研究经四川省妇幼保健院伦理会批准(川妇幼院发〔2019〕128号)。
2.临床检查:对14例患者的临床资料进行回顾性分析,并通过门诊复诊、脑电图(EEG)复查、电话随访等途径获得近期随访资料。所有患者均在医院完成至少头颅影像学CT和(或)MRI检查;完成至少1次16导联4 h左右视频EEG(VEEG)监测,包括完整的清醒-睡眠-觉醒周期记录;所有患儿均完成血常规、肝肾功、甲状腺功能、血气分析,中国-韦氏儿童智力量表(WISC-CR)或Gesell发育商评估,3例完成代谢病血串联质谱及尿液有机酸气相色谱-质谱检测。
3.基因测序:7例患者及父母完成单基因Panel测序,7例患者完成家系全外显子组高通量测序+拷贝数变异(CNV)检查。
结果
1.一般临床资料:本组14例患者,男8例,女6例,男女比例为1.3∶1。14例患儿均有不同程度的全面发育迟缓或智力障碍。4例(P2、P3、P4、P5)韦氏智力评分均为轻度智力障碍,其余10例患者完成Gesell发育商评分,2例(P6、P11)极重度全面发育迟缓,4例(P7、P12、P13、P14)重度全面发育迟缓,4例(P1、P8、P9、P10)中度全面发育迟缓。此外,血常规、肝肾功能、甲状腺功能、血气分析,血串联质谱检测、尿有机酸筛查均未见异常。14例患者的癫痫发作、脑电图特征、病因及神经影像学特征等临床资料详见表1。

图1 P9头颅MRI显示左侧大脑体积较右侧大,白色箭头示脑回增粗,考虑巨脑回畸形
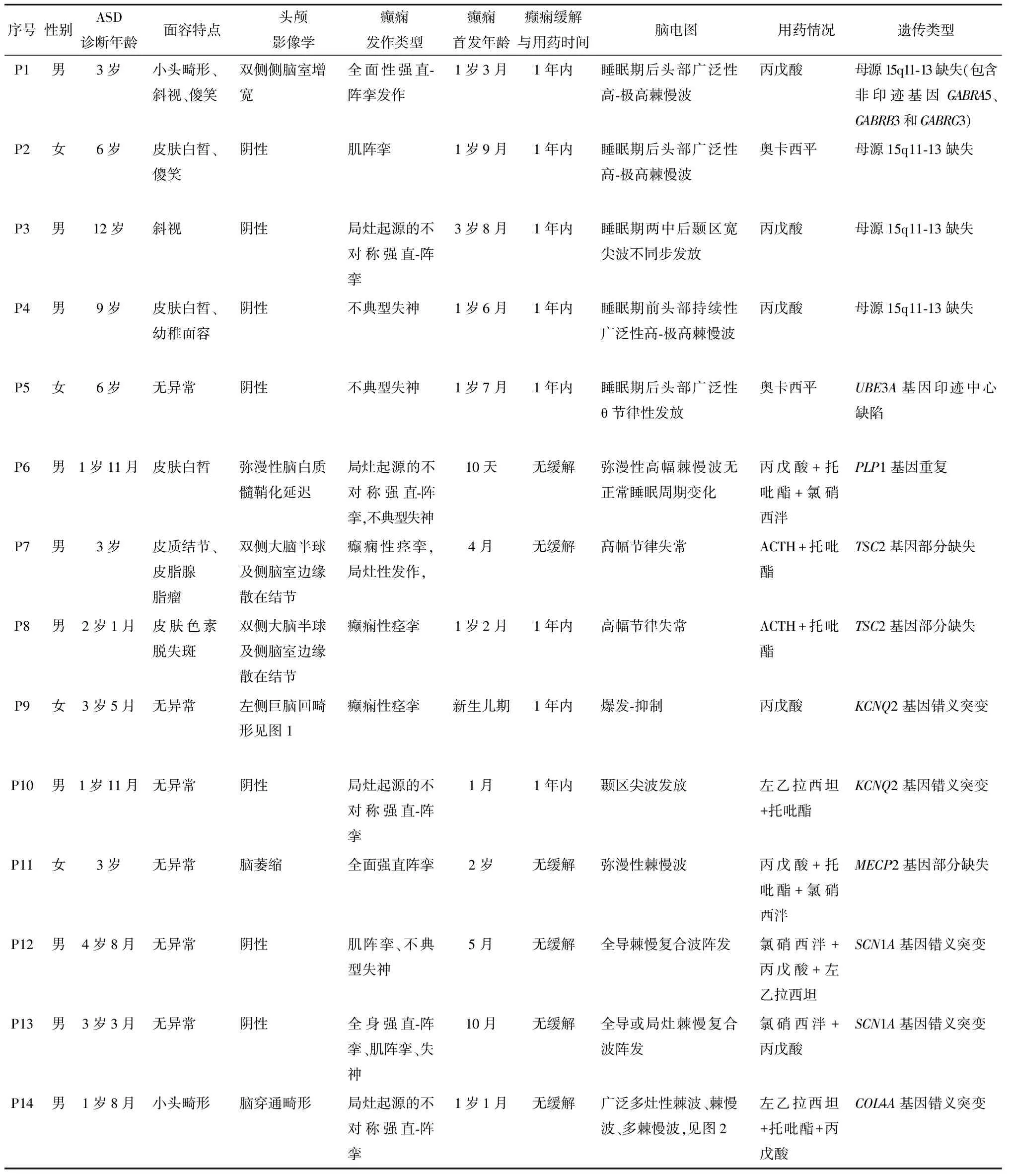
表1 ASD合并癫痫14例患者癫痫临床资料及基因结果汇总
2.基因测序:14例患者中CNVs8例,分别是4例15q11-13缺失(包含非印迹基因GABRA5、GABRB3和GABRG3基因),2例TSC基因部分外显子缺失,1例MECP2基因部分外显子缺失,1例Xq22.2重复(含PLP1基因);单核苷酸变异5例:2例KCNQ2基因错义突变;2例SCN1A基因错义突变,1例COL4A1基因错义突变;DNA甲基化异常1例:UBE3A基因印迹中心缺陷。CNV突变是最常见致病原因,与国外报道一致[3]。P1~P4、P6遗传自无表型母亲,其余9例为新发突变。
3.治疗及随访:所有患者中至少使用过1种抗癫痫药,如丙戊酸、托吡酯、促肾上腺皮质激素(ACTH)、奥卡西平、氯硝西泮。6例(P1~P5、P8)单用丙戊酸或奥卡西平治疗1年内癫痫发作已控制。3例(P7、P9、P10)经2种联合用药1年内癫痫发作控制良好,其余5例家长均对治疗丧失信心,仅愿意继续使用目前抗癫痫药物治疗。P6及P11两例患儿反复感染,P11因感染后气道阻塞死亡,P6因反复呼吸道感染,多次住院于呼吸科及ICU,曾使用有创呼吸机,多种抗癫痫药物治疗惊厥发作无好转,呈非惊厥性癫痫持续状态。
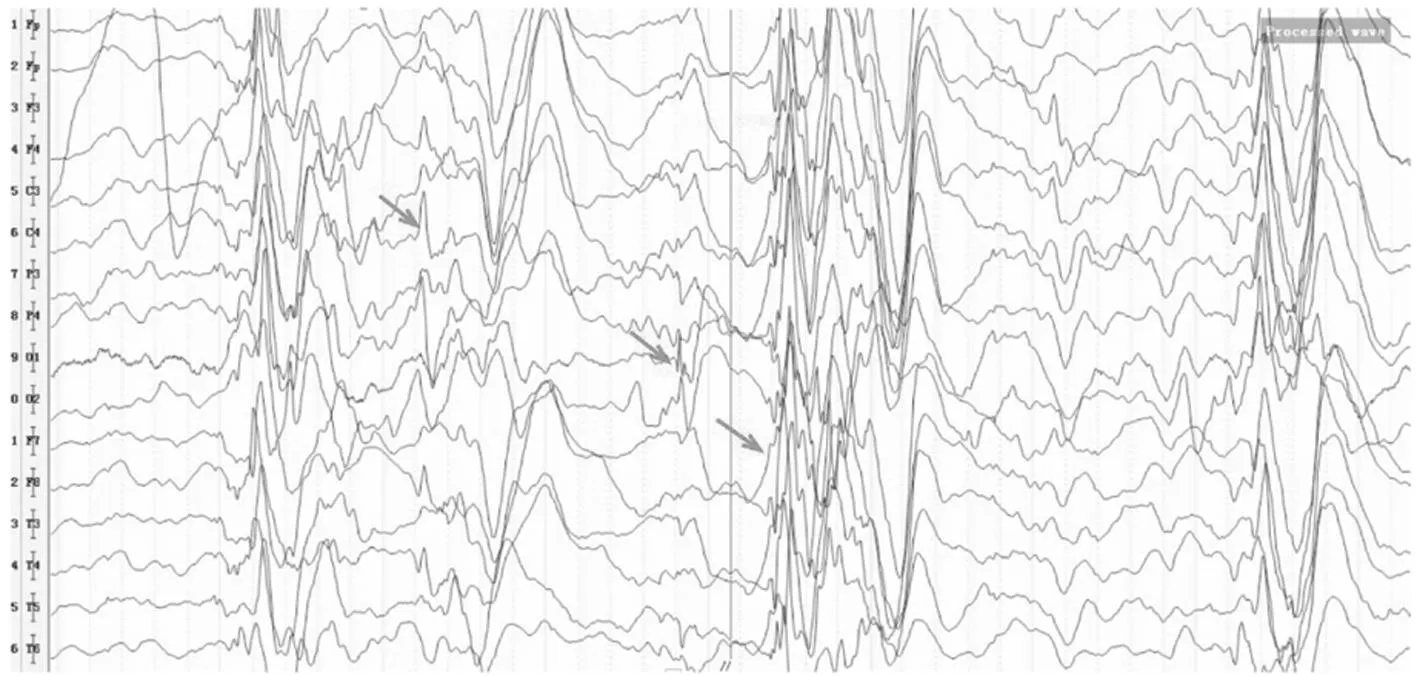
图2 P14患者清醒期脑电图,箭头示双侧弥漫性不规则中高波幅混合漫波、棘波/尖波、棘慢复合波发放
讨论
ASD是一种基于遗传背景及环境因素共同致病的神经发育障碍,其核心症状包括社交缺陷和限制性/重复性行为,严重影响患者与社会互动和沟通,约20%具有可识别的遗传变异[4-5],研究显示同卵双胞胎具有高达70%~90%的一致性致病率[6]。发病机制和临床表现具有异质性。
癫痫是一种大脑神经元异常同步放电的神经系统疾病,大约10%的癫痫发生在生命的前三年[6],其中最严重的形式之一是发育性癫痫性脑病(DEE)。DEE核心内容是在癫痫发作前或发作控制期间独立存在的发育障碍,且发育障碍可能继续进展[7]。
ASD患者伴有癫痫的范围从9%~22%不等,其中癫痫发作有两个高峰:婴儿期和青春期。而既往研究显示癫痫发作的年龄越早,同时合并认知功能障碍的癫痫患儿越容易共患ASD[1]。因此在婴幼儿期出现的DEE是可能合并ASD的高危人群,需要重点监测。这两个疾病之间的关系是双向的,基于目前的假说研究显示,孤独症和癫痫的同时发生,源于与两种疾病相关的共同神经发育通路的损害[8],包括一些参与细胞生长、突触调控、转录调控的生物途径[9]。兴奋/抑制平衡假说认为,GABA受体和谷氨酸受体失活可能破坏了神经回路结构或功能的兴奋性和抑制性,推测这是两种疾病的共同致病机制。这种机制异质性可能反映了不同单基因癫痫性脑病涉及的广泛细胞通路。离子通道病和突触功能障碍是最常见的致病因素。常见的儿童DEE中West综合征、Dravet综合征等共患ASD的患病率较高[9-10]。
ASD还与智力障碍密切有相关[11],ASD合并智力障碍患者中,仅靠CNVs一项检查,阳性率就达到24.4%[8]。既往研究曾显示,智力障碍、性别、年龄及癫痫发作类型是合并ASD的高危因素[12]。但最近研究显示ASD合并DEE患者中,早期治疗与否、智力障碍严重程度以及有效的抗癫痫治疗并不一定能预防共病的发生,因此,这反映了ASD与DEE的遗传异质性[13]。可能需要对大样本进行测序,以增加识别与ASD和DEE相关基因的能力[1]。中国103例癫痫合并孤独症的样本调查分析也支持这一观点,顽固性癫痫发作,孤独症严重情况与智力障碍严重程度之间没有关系[11]。
随着遗传学及基因高通量二代测序的发展,ASD与癫痫之间相重叠的单基因逐渐被发现,如:SCN1A、MECP2、KCNQ2、CDKL5、TSC等基因导致 Dravet综合征、Rett综合征、Angelman综合征(AS)、发育性癫痫性脑病7型、结节性硬化、CDKL5缺乏症等[14]。在部分AS患者中,15q11.2- q13染色体上CNV的缺失,使得UBE3A基因附近的GABAA受体基因(GABRB3、GABRA5和GABRG3)的共同缺失让AS成为ASD与癫痫共病研究的适合对象[15-16]。分析本研究患者基因和可能致病机制,GABRB3、GABRA5、GABRG3、UBE3A基因参与DNA甲基化;MECP2基因参与DNA甲基化与组蛋白修饰,TSC2基因参与调节mTOR信号通路的稳定性,SCN1A基因编码电压门控钠离子通道;KCNQ2基因编码电压门控钾离子通道;COL4A1基因编码IV型胶原α蛋白[17]。
需要注意的是在单基因突变中,SCN1A、KCNQ2、COL4A1基因存在较强遗传异质性前两者基因突变受累的个体临床表现差异大,部分可仅有发热惊厥或良性癫痫表现,无癫痫脑病表现[18-19],COL4A1基因突变患者可有非神经系统异常表现,肾病较为常见[20-22]。对该类变异的患者需详细的询问病史及家族史以做到准确的遗传咨询。
本研究通过对近三年ASD合并癫痫患者临床资料和基因报告的收集,总结了14例遗传相关ASD合并癫痫患者的临床特点及遗传学分析,由于病例数有限,每种基因突变相关的个体病例数更是有限,尚未发现ASD合并癫痫患者表型与基因型之间的联系,下一步可进行蛋白之间联系的研究。由于ASD患者需终生随访,培训和管理,给个人、家庭、社会来带极大的负担,儿童的社会心理干预可改善特定的行为并可能减轻临床症状[22]。如何预防以及在早期高危人群的筛查,以便患者得到及时诊断和治疗,需要儿童神经内科、儿童保健、遗传学医生共同协作,为遗传相关的精准治疗提供帮助。
猜你喜欢
区域治理(2022年40期)2022-11-27 04:01:54
中国民间疗法(2021年5期)2021-06-09 09:21:04
动漫界·幼教365(小班)(2019年10期)2019-10-28 02:04:20
动漫界·幼教365(大班)(2019年10期)2019-10-28 01:54:09
动漫界·幼教365(中班)(2019年10期)2019-10-28 01:53:17
饮食科学(2017年5期)2017-05-20 17:11:53
小天使·二年级语数英综合(2017年4期)2017-04-18 17:29:21
小天使·四年级语数英综合(2017年4期)2017-04-18 09:15:43
西南军医(2015年4期)2015-01-23 01:19:30
少年文艺·开心阅读作文(2014年5期)2014-10-08 16:11:31
