
Characterization of a Novel Esterase Belonging to Family V from Marinobacter flavimaris
2024-03-12 11:14HEJingjingZHANGYunhuiWULeileiWANGYaruZHANGHeLIUZhengangandSHIXiaochong
HE Jingjing, ZHANG Yunhui , WU Leilei, WANG Yaru, ZHANG He,LIU Zhengang, and SHI Xiaochong, ,
1) Frontiers Science Center for Deep Ocean Multispheres and Earth System, and College of Marine Life Sciences,Ocean University of China, Qingdao 266003,China
2) Laboratory for Marine Ecology and Environmental Science, Laoshan Laboratory, Qingdao 266237, China
3) Institute of Evolution & Marine Biodiversity, Ocean University of China, Qingdao 266003, China
Abstract Lipolytic enzymes have attracted enormous attentions because of their ability in ester hydrolysis, ester synthesis, transesterification and other biochemical reactions. Bacteria are important sources of lipolytic enzymes applied in industry. Here, a novel lipolytic enzyme encoded by esterase gene est1347 was identified in Marinobacter flavimaris WLL162, and was purified and characterized. The lipolytic enzyme Est1347 consisted of 312 amino acid residues and a 21-amino-acids N-terminal signal peptide with a predicted molecular weight of 34.2 kDa. It belongs to family V of bacterial lipolytic enzymes based on the amino acid sequence homology analysis. Est1347 is a mesophilic and alkali-resistant enzyme with the highest activity at 45℃ and pH 8.5; it is stable at temperatures below 50℃ and pH 7.5 – 11.0. Est1347 showed a preference for middle-length chain substrate p-NPC10 and a wide range of other substrates. The Km, Vmax, Kcat and Kcat/Km values of Est1347 for p-NPC10 in pH 8.5 at 45℃ were 0.9411 mmol L−1, 1285 μmol min−1 mg−1, 698.91 s−1 and 743.65 s−1 (mmol L−1)−1, respectively. It is also tolerant to the metal ions, organic solvents and detergents. In conclusion, the esterase Est1347 laid a foundation for further study of bacterial lipolytic enzyme family V.
Key words esterase; Marinobacter flavimaris; enzymatic properties; lipolytic enzyme family V
1 Introduction
Lipolytic enzymes can catalyze the hydrolysis and synthesis of the ester bond, and have a function in transesterification and other reactions. It distributes extensively in bacteria, archaea, viruses and eukaryote organisms, while bacteria are the main sources of industry-applied lipolytic enzymes. Bacterial lipolytic enzymes include carboxylesterases (EC 3.1.1.1, esterase) and true lipases (EC 3.1.1.3, lipase), and have putative physiological functions with virulence, metabolism and growth promotion (Kovacicet al.,2019). Esterases prefer water-soluble short-chain to medium-chain esters, whereas lipases hydrolyze the water-insoluble long-chain triglycerides (Jaegeret al., 1994). The catalysis of lipolytic enzymes is characterized by mild reaction conditions, high efficiency and no need for coenzyme, which can be widely applied to food, pharmaceutical and cosmetic industries (Bornscheuer, 2002).
Bacterial lipolytic enzymes are classified into eight families (Family I – VIII) based on the amino acid sequences similarities and physiological properties (Arpigny and Jaeger, 1999). Recently, an increasing number of lipolytic enzymes have been characterized and classified, while 35 esterase families and 11 true lipase families have been newly introduced (Hitch and Clavel, 2019). The catalytic domain of lipolytic enzymes is composed of a classic α/βhydrolase fold, with eight β-sheets connected by α-helices in the central position and α-helices flanked on both sides(Mølgaardet al., 2000). The catalytic triad Ser, Asp/Glu and His are located in the catalytic domain and the active site Ser is in the conserved GXSXG pentapeptide (Olliset al.,1992). According to the structural homology, the ESTHER database reclassifies proteins containing the α/β-hydrolase fold into four blocks: C, H, L and X. Family V is classified into block X and it is divided into three subfamilies: Family V.1, Family V.2, Family V.3 (Lenfantet al., 2012).
Microorganisms from extreme environments are found to possess lipolytic enzymes with special characteristics and these lipolytic enzymes can be used as uniquely valuable biocatalysts (Vermaet al., 2021). A thermostable lipolytic enzyme PLP from the metagenomic library of deep-sea hydrothermal vent demonstrates optimal activity at 70℃ and this property implies it can be used in the high-temperature food processing industry (Fuet al., 2015). The hyperthermostable carboxylesterase EstD9 from thermophilicAnoxybacillus geothermalisD9 presents excellent stability at high-temperature range (70 – 100℃) and high tolerance to various surfactants and metal ions; these enzymatic features suggest it is a highly suitable catalyst for industrial bioprocesses (Johanet al., 2022). The cold-adapted esterase EstDR4 from extremophileDeinococcus radioduransdisplays regioselectivity, enantioselectivity, and degradation effects on four insecticides, which may have the potential to be used in the pharmaceutical industry and the treatment for pollution caused by insecticides (Zhanget al.,2021). Another psychrophilic esterase Est19 from the Antarctic bacteriumPseudomonassp. E2-15 shows the stereo-specificity that can identify the methyl-L lactate and methyl-D lactate, and this characteristic suggests Est19 may be useful for cold and chiral catalyses (Liuet al., 2021).Additionally, the halophilic esterase EstA1 isolated from the marine bacteriumAlteromonassp. 39-G1 has excellent salttolerance characteristic and its activity is continuously increased with increasing NaCl concentration, and this unique property enhances its academic and industrial values(Wonet al., 2020). Another novel high salt-tolerant esterase EstSHJ2 remains high activity in the solution with 1.0 – 3.5 mol L−1NaCl while the activity is very low without NaCl(Wanget al., 2020). It is previously reported some properties of halophilic lipase can improve the flavor of fish sauce(Kanlayakrit and Boonpan, 2007), so the study of the halophilic lipolytic enzyme may have applicable values in the food industry. The lipolytic enzymes with diverse enzymatic characteristics from different environments have important applications in the food, dairy, detergent and pharmaceutical industries (Fuciñoset al., 2012). Therefore, mining unique esterase resources has raised enormous attention.
Marinobacterbelongs toAlteromonadaceaein the class ofγ-Proteobacteria, which can degrade hydrocarbon and utilize the lipid (Duran, 2010; Boninet al., 2015). It is previously reported thatMarinobacterplays an indispensable role in the remediation of hydrocarbon pollution in highsalt environments (Lyuet al., 2022; Zhanget al., 2022).Meanwhile, mostMarinobacterisolates are found to have halophilic lipolytic ability, such asMarinobacter lipolyticusCEES 33 (Oveset al., 2017),Marinobacter litoralisSW-45 (Musaet al., 2019) andMarinobacter lipolyticusSM19 (Martínet al., 2003). Therefore, exploring the potential lipolytic enzymes inMarinobacteris of great significance. The halophilic lipase, LipBL, isolated from theMarinobacter lipolyticusSM19, belongs to family VIII and is highly efficient in producing eicosapentaenoic acid (EPA),whose properties are of great interest in the food industry(Pérezet al., 2011). The active sites of LipBL are investigated through site-directed mutagenesis, the critical S-XX-K motif is found to have a crucial effect on the catalysis and the conserved GXSXG pentapeptide presumably represents a substrate-binding site, which is different from the classical catalytic residues (Pérezet al., 2012).
Some reported lipolytic enzymes have poor stability,which is an important factor limiting their industrial application. Therefore, exploring novel lipolytic enzymes with great stability from microorganisms is a feasible method.Recently,Marinobacter flavimarisWLL162 with strong lipolytic enzyme activity was isolated from the Western Pacific Cobalt-rich crust (Wu, 2020) and 12 putative lipolytic enzyme genes were identified in its genome sequence.In this study, a novel esterase geneest1347fromM. flavimarisWLL162 was cloned and the esterase Est1347 was expressed, purified and characterized. These results propose a novel esterase from the marine source and provide a supplement for family V of the bacterial lipolytic enzyme.
2 Materials and Methods
2.1 Bacterial Strains, Plasmids, Media and Growth Conditions
Marinobacter flavimarisWLL162 (=Marinobacter flavimarisSW-145T) (Yoonet al., 2004) was isolated from the sediment (5443 m water depth) of Western Pacific Cobalt-rich crust area at 157.2144˚E, 19.60048˚N and cultured using 2216E (MA, marine agar) at 28℃. The cloning and expression vectors were pUCm-T (Sangon Biotech,Shanghai, China) and pET-24a (+) (Novagen, Beijing, China), respectively.Escherichia coliDH5α was used for gene clone andE. coliBL21 (DE3) was used for the protein expression, while both of them were cultured at 37℃ in Luria-Bertani (LB) agar.
2.2 Sequence and Phylogenetic Analysis of Esterase Gene
The genomic DNA from WLL162 was extracted using a DNA extraction kit (TIANGEN BIOTECH, Beijing, China). The sequencing and assembling of total genomic DNA were based on the methods of Linet al. (2018). The prediction and annotation of the Open reading frame (ORF)were carried out by the RAST server (Overbeeket al., 2014).The amino acid sequence was analyzed by BLASTP against the protein data bank (PDB) database and UniProt Knowledgebase (UniProtKB) (Batemanet al., 2021). The multiple sequence alignment between Est1347 and reference sequences was performed using ClustalX 2.1 (Larkinet al.,2007) and the result was rendered using the ESPript 3.0 online tool (ESPript 3.x / ENDscript 2.x (ibcp.fr)). The phylogenetic tree based on the amino acid sequence was constructed by MEGA X (Kumaret al., 2018), with 1000 bootstraps, and sequences of other lipolytic enzymes were downloaded from GenBank. The three-dimensional model was predicted using Swiss-Model online tool and analyzed by PyMOL. The molecular weight and pI of esterase were predicted by the ExPASy database (Gasteigeret al., 2003)and the signal peptide was predicted by the SignalP 5.0 server (Almagro Armenteroset al., 2019).
2.3 Cloning, Expression and Purification of Est1347
To obtain the recombinant proteins without signal peptide, theest1347gene (GenBank accession number OQ29 5858) was amplifiedviapolymerase chain reaction (PCR)with the primer pairs,est1347-F(CGCATGTG CTCTCGTCAG) andest1347-R(CCCCTCGA CCGGCTGCC), which containedBamHI andHindⅢ digestion sites (underlined), respectively. The PCR products were cloned to the pUCm-T vector and the resulting plasmid was transformed intoE. coliDH5α competent cells.The recombinant plasmid pUCm-T/est1347was extracted and digested byBamHI andHindⅢ restriction endonucleases after sequencing confirmation; the products were purified and inserted into the linearized plasmid pET-24a (+).The correct expression vector pET-24a (+)/est1347was introduced intoE. coliBL21 (DE3) competent cells for protein expression. The recombinant cells were grown in LB medium supplemented with 50 μg mL−1kanamycin at 37℃.At the mid-exponential growth phase (OD600nm= 0.4 – 0.6),isopropyl-β-D-thiogalactopyranoside (IPTG) was added to a final concentration of 0.05 mmol L−1, and the cultivation was continued at 16℃, with shaking at 150 r min−1for another 12 – 14 h. The induced cells were harvested by centrifugation at 8000 r min−1, 4℃ for 10 min, washed with prechilled PBS buffer, and resuspended with prechilled binding buffer (20 mmol L−1Tris-HCl, 0.5 mol L−1NaCl, pH 8.0).After ultrasonic disruption on ice, the cell debris was removed by centrifugation at 12000 r min−1, 4℃ for 10 min.The supernatant was filtered through a 0.22-μm-pore-size filter and loaded onto the Ni-NTA affinity column (Qiagen,Hilden, Germany). Proteins were purified according to the manufacturer’s recommendations. The eluate containing the target protein was dialyzed with dialysate (20 mmol L−1Tris-HCl, 0.85% NaCl, 10% glycerol, pH 8.0) for 24 h at 4℃. The expression and purification of protein were verified by 12% sulfate-polyacrylamide gel electrophoresis(SDS-PAGE) (Laemmli, 1970).
2.4 Esterase Activity Detection of Est1347
The enzymatic activity of purified recombinant Est1347 was tested by the reported method with some modification(Wanget al., 2016). Esterase activity againstp-nitrophenyl (p-NP) esters was determined by measuring the amount ofp-nitrophenol released by catalytic hydrolysis. The production ofp-nitrophenol was measured by the absorbance at 405 nm. The standard reaction system contained 100 m mol L−1ofp-nitrophenyl butyrate (p-NPC4) in acetonitrile,ethanol, and 50 mmol L−1of Tris-HCl buffer (pH 8.5) at a ratio of 1:4:94 (V/V/V). The reaction system was preincubated at 45℃ for 5 min and initiated by adding 10 μL of purified enzyme solution to 1 mL of substrate mixture. After 10 min, the absorbance of the reaction system at 405 nm was measured. One unit of esterase activity was defined as the amount of esterase capable of releasing 1 μmol ofp-nitrophenol per minute. Protein concentration was measured by the Bradford method (Bradford, 1976) using bovine serum albumin (BSA) as the standard.
2.5 Biochemical Characterization of Est1347
The optimal temperature of Est1347 was determined in optimal pH at 0, 5, 15, 20, 30, 40, 45, 50, 55, 60, and 70℃.For detecting the thermostability, the enzyme samples were incubated at 0, 5, 15, 20, 28, 40, 45, 50, 55, 60, 70, 80,and 90℃ for 1 h, and then the residual activities were measured. The optimal pH of Est1347 was measured with pH range of 3.0 – 11.0 using four kinds of buffer systems: 50 mmol L−1citric acid/sodium citrate buffer (pH 3.0 – 6.0), 50 mmol L−1Na2HPO4-NaH2PO4buffer (pH 6.0 – 8.0), 50 mmol L−1Tris-HCl (pH 7.5 – 9.0) and 50 mmol L−1Na2HPO4-NaOH (pH 10.0 and 11.0). To determine the pH stability of esterase, the purified enzyme samples were preincubated in the above-mentioned buffers at 4℃ for 12 h,and the residual activities were measured at 45℃. Substrate specificity was assayed with the followingp-nitrophenyl esters:p-nitrophenyl acetate (p-NPC2),p-nitrophenyl butyrate (p-NPC4),p-nitrophenyl caproate (p-NPC6),p-nitrophenyl caprylate (p-NPC8),p-nitrophenyl decanoate (p-NPC10),p-nitrophenyl laurate (p-NPC12) andp-nitrophenyl palmitate (p-NPC16).
The kinetic parameter of esterase was determined by measuring enzymatic activity usingp-NPC10 as substrate at 45℃ in pH 8.5 for 10 min with substrate concentrations of 0.01, 0.05, 0.1, 0.15, and 0.2 – 1.2 mmol L−1(at an interval of 0.2). TheKmandVmaxvalues were calculated using the Michaelis-Menten equation in GraphPad Prism 9.0. And theKcatandKcat/Kmwere calculated based on the value ofVmax.
The effect of metal ions (Fe3+, Co2+, Mg2+, Cu2+, Ni2+,Mn2+, Na+, K+, Ca2+, Zn2+) and metal ion chelator EDTA on esterase activity was determined by adding different ions and EDTA at a final concentration of 1 mmol L−1and 10 mmol L−1into the reaction system. The effect of salt concentration on Est1347 activity was measured with the concentration of NaCl of 0 – 5 mol L−1in the reaction system.The organic solvents (acetonitrile, DMF, isopropanol, methanol, acetone, DMSO, glycerol) were added respectively to the standard reaction system (10%, V/V) to investigate the effect of organic solvents on Est1347. The detergents (SDS, Triton X-100, Tween-20, Tween-80) were added respectively to the standard reaction system (1%, V/V)to study the effect of detergents on the enzyme.
3 Results
3.1 Protein Sequence and Phylogenetic Analysis of Est1347
Based on the result of genome annotation of the putative esterase geneest1347, a 939-bp open reading frame(ORF) was identified. The ORF encodes a protein containing 312 amino acid residues with a predicted molecular weight of 34.2 kDa and a predicted pI of 4.51. The N-terminal signal peptide was composed of 1 – 21 amino acids.Sequence alignment revealed that this protein shared the highest identity of 41.42% with the membrane phospholipase A (UniProtKB: Q9KJG6) fromPseudomonas aeruginosaPAO1. The phylogenetic analysis of Est1347 and known bacterial lipolytic enzyme families showed that Est1347 belongs to family V of bacterial lipolytic enzymes(Fig.1). Multiple sequence alignment of Est1347 with highly related homologs showed that the canonical catalytic triad containing Ser139, Asp261and His289, and Ser139is located in the GXSXGG motif (Fig.2). A three-dimensional structure of Est1347 was predicted based on the template 6I8W (the crystal structure of the protein Q9KJG6) and the model structure revealed that Est1347 had a typical α/β-hydrolase fold (Fig.3a). The multiple sequence alignment and phylogenetic analysis results confirmed that Est1347 is a novel lipolytic enzyme belonging to bacterial lipolytic enzyme family V.

Fig.1 Phylogenetic tree of lipolytic enzymes based on the homology of protein sequences, constructed using the neighborjoining method by MEGA X. The sequences used in the analysis were obtained from the GenBank database. Bootstrap values are based on 1000 replicates, and only values > 50% are shown. The scale bar indicates the number of amino acid substitutions per site.

Fig.2 Multiple sequence alignments of amino acid sequences of Est1347 and homologs from different bacteria. Accession numbers of proteins in the UniProtKB are given for Q9KJG6 (Pseudomonas aeruginosa PAO1), P24640 (Moraxella sp.TA144), Q02104 (Psychrobacter immobilis B10), Q1QEU6 (Psychrobacter cryohalolentis K5), B0V9K7 (Acinetobacter baumannii AYE). The active-site residues are indicated by circles below the sequences.
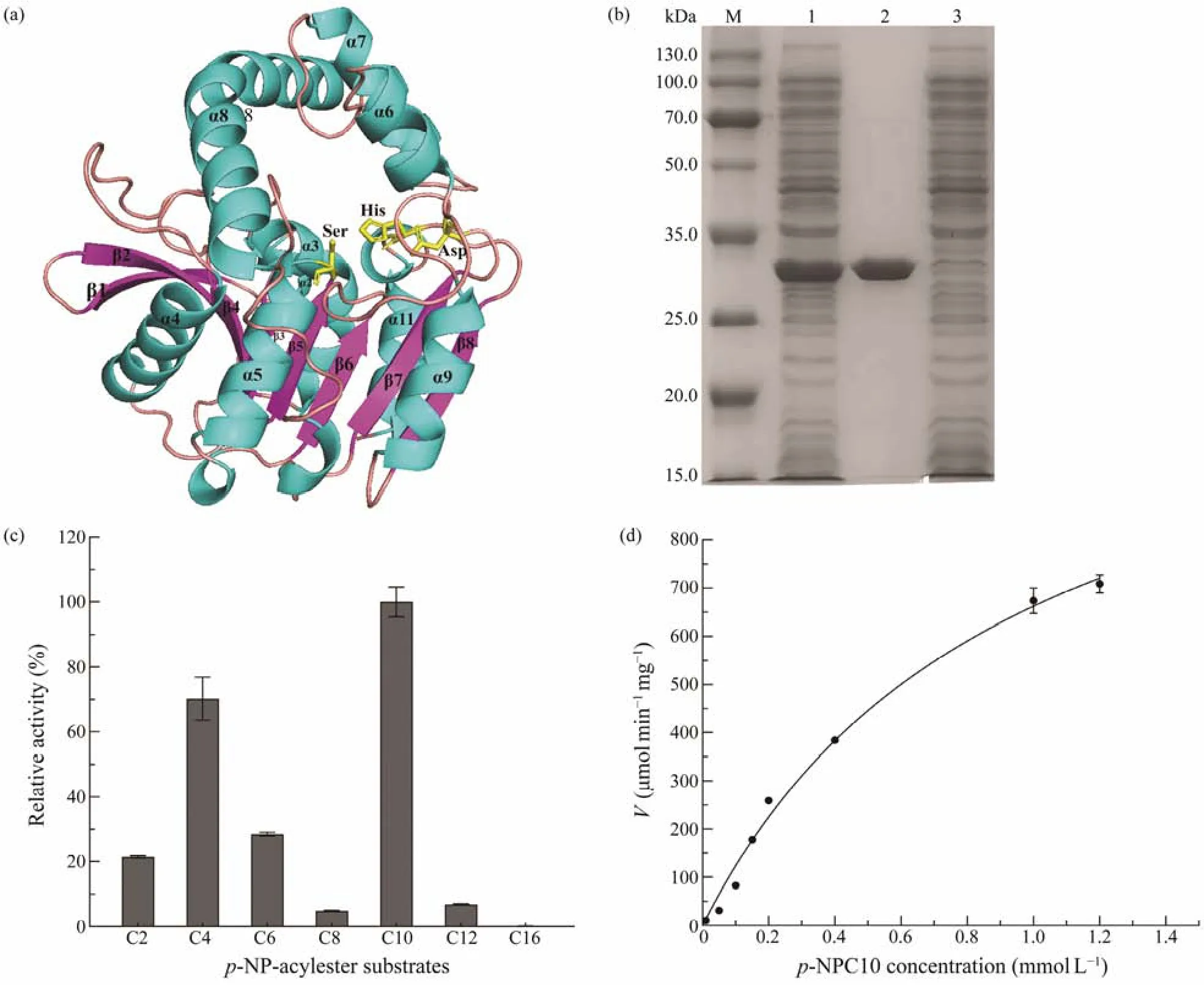
Fig.3 Predicted three-dimensional structure of Est1347 (a); SDS-PAGE analysis of recombinant esterase Est1347 (b); substrate specificity of Est1347 (c); and enzyme kinetics of recombinant Est1347 (d). (a) The three-dimensional structure of Est1347 was modeled against template 6I8W (the crystal structure of the protein Q9KJG6). The catalytic triad Ser, Asp and His is shown in yellow sticks. (b) SDS-PAGE of recombinant esterase Est1347. Lanes: M, molecular mass markers;lane 1, cell-free extracts of E. coli BL21 (DE3) harboring plasmid pET-24a (+)/est1347; lane 2, purified Est1347; lane 3,cell-free extracts of E. coli BL21 (DE3) harboring plasmid pET-24a (+). (c) Substrate specific of Est1347 toward p-NP esters. Substrate specific was determined using p-NP esters with various chain lengths (C2 – C16) at a final concentration of 1 mmol L−1 at 45℃ in 50 mmol L−1 Tris-HCl buffer (pH 8.5). (d) Determination of enzyme kinetics of Est1347 toward p-NPC10. All data are shown as mean ± SD (n = 3).
3.2 Expression and Activity of Esterase Est1347
The vector pET-24a (+) was used to express the esterase in theE. coliBL21(DE3) cells. The purified recombinant proteins were evaluated by SDS-PAGE (Fig.3b),which displayed a single band with an apparent molecular weight of approximately 32 kDa, accordant to the predicted molecular mass (32.02 kDa). About 7 mg of the recombinant protein was obtained after His-tag affinity purification from 300 mL LB broth. Est1347 could efficiently hydrolyze short- to middle-lengthp-NP esters (C2 – C10) with the highest activity towardp-NPC10 (6991 U mg−1) followed byp-NPC4 (4927.3 U mg−1) andp-NPC6 (2036.0 U mg−1)among thep-NP esters examined (Fig.3c). The specific activity of Est1347 was observed usingp-NPC10 as substrate in 50 mmol L−1Tris-HCl (pH 8.5) at 45℃, and theKmandVmaxfor esterase were 0.9411 mmol L−1and 1285 μmol min−1mg−1(Fig.3d), respectively. TheKcatandKcat/Kmvalues of Est1347 were calculated as 698.91 s−1and 743.65 s−1(mmol L−1)−1, respectively. In addition, Est1347 also exhibited hydrolytic activity against tributyrin and Tween-20, 40 and 80 (Fig.4).
3.3 Effects of Temperature and pH on Activity and Stability of Est1347
The purified esterase was evaluated for its activity and stability at different temperatures and pH values. The optimum temperature of Est1347 was 45℃ and it was active in a wide range of 0 – 60℃ (Fig.5a). For the stability of Est1347, it could retain more than 80% of its highest activity after 1 h incubation at temperatures 0 – 50℃, but lost almost all activity after 1 h incubation at 90℃ (Fig.5b).

Fig.5 Responses of temperature of recombinant esterase Est1347. (a) Effect of temperature on Est1347 activity. The highest activity of Est1347 at 45℃ was taken as 100%. (b) Thermostability of Est1347. The enzyme was incubated at 0 – 90℃for 1 h. The remaining activity was measured under optimal conditions. The highest activity at 15℃ was taken as 100%.The data are shown as mean ± SD (n = 3).
Est1347 showed the highest activity at pH 8.5 and exhibited activity in a narrow pH range (6.0 – 9.0, Fig.6a). Moreover, it could maintain most of its initial activity after 12 h incubation at pH 7.5 – 11.0 (Fig.6b), which indicated that Est1347 had relatively high pH stability and it preferred alkaline environment.

Fig.6 Responses of pH of recombinant esterase Est1347. (a) Effect of pH on the activity of Est1347. The absorbance of the reaction system was measured at 405 nm. (b) pH-stability of Est1347. The enzyme was incubated at pH from 3.0 to 11.0 for 12 h at 4℃. The remaining activity was measured under optimal conditions. The absorbance of the reaction system with preincubated enzyme was measured at 405 nm. The data are shown as mean ± SD (n = 3).
3.4 Effects of Metal Ions, EDTA and Salinity on Activity of Est1347
The effects of metal ions and ion chelator EDTA on the activity of Est1347 were also investigated (Fig.7a). The results showed that only K+(1 mmol L−1) could improve the enzyme activity. Mg2+, Mn2+, Na+, Ca2+(1 mmol L−1and 10 mmol L−1) and K+(10 mmol L−1) had no obvious effect on Est1347. Fe3+, Co2+, Cu2+, Ni2+and Zn2+(1 mmol L−1and 10 mmol L−1) could inhibit the activity of Est1347 significantly. The ion chelator EDTA had no significant effect on Est1347 at 1 mmol L−1while inhibited enzymatic activity at 10 mmol L−1.

Fig.7 Response of Est1347 to metal ions and EDTA (a) and salinity (b). (a) Effects of 1 mmol L−1 and 10 mmol L−1 metal ions and EDTA on the activity of Est1347; ** P < 0.01, * P < 0.05; (b) Effects of different NaCl concentrations on the activity of Est1347. The data are shown as mean ± SD (n = 3).
The activity of Est1347 decreased (Fig.7b) with the increase of salinity. Est1347 could remainca.83% andca.69% of the initial activity with 0.5 mol L−1and 1 mol L−1NaCl in the reaction system respectively, which indicates that Est1347 is a moderate salt-tolerant esterase.
3.5 Effects of Organic Solvents and Detergents on the Activity of Est1347
The effects of different organic solvents on the activity of Est1347 were measured (Fig.8). The methanol, DMSO and glycerol had no obvious effect on the enzymatic activity of Est1347. While acetonitrile, DMF, isopropanol, ethanol and acetone inhibited the enzymatic activity significantly, and acetonitrile showed the strongest inhibition effect which could reduce the activity of Est1347 to onlyca.16%.The detergents also showed significant effects on the activity of Est1347 (Table 1). The ionic detergent SDS and nonionic detergents Tween-20 and Tween-80 inhibited the activity of Est1347, wherein SDS and Tween-80 could inhibit the activity completely. However, the non-ionic detergent Triton X-100 could increase the enzyme activity to 106.9%.

Table 1 Effects of various detergents on the activity of Est1347

Fig.8 Response of Est1347 to various organic solvents. The value observed without organic solvent was taken as control (100%). ** P < 0.01, * P < 0.05.
4 Discussion
In this study, a novel esterase Est1347 was identified from the marine bacteriumMarinobacter flavimarisWLL162 and classified into the bacterial lipolytic enzyme family V based on the conserved motif and homologs of amino acid sequences. Among the characterized lipolytic enzymes,Est1347 had the highest identity (41.24%) to the α/β fold hydrolase ofPseudomonas aeruginosaPAO1 (Bleffertet al.,2022), which was a novel bacterial virulence factor and the esterase activity of which was characterized using 4-methylumbelliferyl palmitate andp-nitrophenyl butyrate as substrates. Est1347 also shared similarities with esterase EstPc fromPsychrobacter cryohalolentisK5 (accession number ABE73807.1), lipase Lip1 fromPsychrobacter immobilisB10 (accession number CAA47949.1) and lipase Lip3 fromPsychotroph moraxellaTA144 (accession number CAA37863.1), with 36.88%, 35.21% and 34.86% amino acid sequence identity, respectively. These three lipolytic enzymes were characterized by psychrophilic activeties (Felleret al., 1991; Arpignyet al., 1993; Novototskaya-Vlasovaet al., 2012). However, Est1347 showed the highest activity at moderate temperature (Table 2), which indicates that Est1347 probably represents the moderate temperature type esterase in family V.

Table 2 Comparison of properties of known esterases from different bacterial lipolytic enzyme families
Lipolytic enzymes grouped in family V were originated from various organisms in different environments and showed diverse characteristics, they remained to be further explored (Ruizet al., 2007). The novel esterase Est1347 in this study displayed great thermostability at its optimal temperature range (retaining over 80% activity after incubation at 0 – 50℃ for 1 h). The esterase Est_p1, a mesophilic enzyme and a member of family V, displayed the highest activity at 40℃ and retained about 50% of activity after incubated at 40℃ for 1 h while the residual activity at 45℃or 50℃ dropped rapidly within 15 min (Penget al., 2011).Another esterase EstIM1 showed its highest activity at 40℃ but it was sensitive to 40℃, whose activity could retain no more than 20% after incubation at 40℃ for 20 min(Koet al., 2012). These results indicate Est1347 is more suitable for medium-temperature catalysis and is capable of maintaining catalytic activity at the optimum reaction temperature for a long time.
For the optimal pH and pH stability, the maximum activity of Est1347 was observed at pH 8.5, similar to other alkaline esterases belonging to family V, which include EstPc fromPsychrobacter cryohalolentisK5 (Novototskaya-Vlasovaet al., 2012), Est16 from a metagenomic library of a microbial consortium specialized for diesel oil degradation (Pereiraet al., 2015) and EstKa fromKlebsiella aerogenesGF-0526 (Gaoet al., 2021). Interestingly, Est1347 retained most of the enzyme activity at pH 7.5 – 11.0 after incubation at relevant buffers for 12 h, which indicated it had obvious advantages in the preparation of enzyme and industrial application. Alkali-stable enzymes usually possess more arginine and neutral hydrophilic amino acid (histidine, asparagine and glutamine) residues, and the arginine residues can form hydrogen bonds or ion pairs to adapt to the alkaline conditions (Shiraiet al., 1997). Est1347 had a proportion of 15.4% of arginine and neutral hydrophilic amino acid residues, which may be related to its alkali stability.
Most lipolytic enzymes in family V preferred the shortor middle-length (C < 10) chainsp-NP esters as the substrate. For example, EstV showed a preference for shortlength (C2 – C6)p-NP esters (Ruizet al., 2007), est-OKK had the maximal activity towardp-NPC4 (Yanget al., 2018),BroH displayed the highest activity towardp-NPC6 (Chenet al., 2013) and FCLip1 preferentially hydrolyzedp-NPC8(Caiet al., 2011). However, Est1347 had the highest activity againstp-NPC10, which was different from other members and provided new insight into the lipolytic enzyme family V. According to former reports, the substrate-binding pocket of theE. colithioesterase TesA was reshaped by structure-guided mutational screening to improve the C8 substrate selectivity (Denget al., 2020). Considering the preference of Est1347 to substratep-NPC10, we deduced the substrate-binding pocket of Est1347 probably prefer-red binding the C10 substrate over the C4 substrate. This deduction requires further confirmation. Furthermore, Est1347 prefers short- to middle-length chain acyl substrates and could degrade tributyrin, which confirms the function of Est1347 as an esterase. In addition, Est1347 also could degrade non-ionic detergents Tween-20, Tween-40 and Tween-80, indicating Est1347 might be employed in detergent pollution treatment.
The specific activities of enzymes from different sources were summarized and the kinetic paraments of Est1347 were compared with other esterases. The best substrate of Est1347 wasp-NPC10 and theVmaxvalue (1285 μmol min−1mg−1) of Est1347 againstp-NPC10 was at a moderate level (Table 2). It had been reported that medium chain length(C6 – C10) fatty acids and their derivatives had unique application values, such as antibacterial, lubricant, substitutes for types of gasoline, and intermediates for herbicide synthesis (Sarriaet al., 2017). The property of Est1347 which preferred hydrolyzingp-NPC10 implied it might have the potential in the production of middle-chain fat acid. Besides,the cleaning experiment was also performed according to the method of Zafaret al.(2022). And esterase Est1347 combined with detergent could removeca. 14.12% more stained oil than detergent alone. The oil in this study was with relatively long chain length. Est1347 possibly has higher cleaning efficiency on the middle-length oil stain. The potential of Est1347 applied to commercial laundry detergents still needs to be further investigated.
5 Conclusions
In this study, a novel esterase geneest1347was identified and cloned fromMarinobacter flavimarisWLL162 and the encoding protein Est1347 was expressed successfully usingE. coli. The esterase Est1347 belongs to the bacterial lipolytic enzyme family V and is a mesophilic and alkali-resistant enzyme. Est1347 showed a preference for middle-length chain substratep-NPC10 and exhibited a large range of substrates. Furthermore, Est1347 showed great thermostability and pH stability, which could maintain more than 80% of its maximal activity at 0 – 50℃ and pH 7.5 –11.0. These characteristics of Est1347 prompted it to be a candidate for biotechnological applications in industry.
Acknowledgements
This work was supported by the projects from the National Natural Science Foundation of China (No. 42230411),and the China Ocean Mineral Resources R and D Association (COMRA) Special Foundation (No. DY135-B2-10).
 Journal of Ocean University of China2024年1期
Journal of Ocean University of China2024年1期
- Journal of Ocean University of China的其它文章
- Using Natural Radionuclides to Trace Sources of Suspended Particles in the Lower Reaches of the Yellow River
- Eutrophication of Jiangsu Coastal Water and Its Role in the Formation of Green Tide
- Evaluation of the Shallow Gas Hydrate Production Based on the Radial Drilling-Heat Injection-Back Fill Method
- Microstructure Characterization of Bubbles in Gassy Soil Based on the Fractal Theory
- Morphological and Sulfur-Isotopic Characteristics of Pyrites in the Deep Sediments from Xisha Trough, South China Sea
- Deformation Characteristics of Hydrate-Bearing Sediments