
Transcriptome Analysis of Heterosis in Survival in the Hybrid Progenies of ‘Haida No. 1’ and Orange-Shelled Lines ofthe Pacific Oyster Crassostrea gigas
2024-03-12 11:14YANGHangandLIQi
YANG Hang, and LI Qi
Key Laboratory of Mariculture, Ministry of Education, Ocean University of China, Qingdao 266003, China
Abstract Heterosis has been exploited to enhance the yield and adaptability in various shellfish species; however, the molecular basis of it remains unclear. The Pacific oyster Crassostrea gigas is one of the most economically important aquaculture species, and its productive traits can be improved by hybridization. Here, an intraspecific cross between orange shell (O, 10th generation) and‘Haida No. 1’ (H, 13th generation) of C. gigas was performed to assess the heterosis of survival trait. Survival rates of hybrid family(OH) and inbred families (HH and OO) were compared at larval stage, and eyed-pediveliger larvae of three families were subjected to transcriptome analysis. The analysis results of best-parent heterosis and mid-parent heterosis showed that the hybrid family exhibited a high heterosis in survival relative to the parental families. The OH-M (OH vs. OO) and OH-P (OH vs. HH) had 425 and 512 differentially expressed genes (DEGs), respectively. Functional enrichment analysis of these DEGs revealed that the significantly enriched genes function in virion binding, C-type lectin receptor signaling pathway, cellular defense response and other immune-related processes, which involves perlucin-like protein, CD209 antigen-like protein, ZNFX1, caspase-3 and acan genes. These differentially expressed genes in OH-M and OH-P, together with the immune-related processes mentioned above may play an important role in the larval survival of C. gigas. In addition, three genes (CYP450, fucolectin and perlucin-like) are associated with the orange shell and low survival of maternal oyster OO. These findings provide support for the application of hybrid with superior survival and will facilitate the understanding of heterosis formation in the Pacific oyster.
Key words Crassostrea gigas; survival rate; heterosis; transcriptome
1 Introduction
Heterosis, or hybrid vigor, is the phenomenon whereby heterozygous F1 produced by different genetic parents has superior performance in growth, survival and resistance to stress than parental homozygote or inbred lines (Birchleret al., 2010; Songet al., 2010). Due to known biological and economic precedence of heterosis, the research community has been fascinated by the underlying molecular mechanisms for over a century. Several genetic hypotheses(i.e., dominance, overdominance, pseudo-overdominance and epistasis hypothesis) were proposed to explain heterosis (Bruce, 1910; Lippman and Zamir, 2007; Shanget al.,2016), while a single genetic mechanism cannot fully explain the mechanism (Shahzadet al., 2020).
With the rapid advancements of molecular biological approaches, molecular evidence underlying heterosis has begun to be elucidated. At present, cDNA-amplified fragment length polymorphism, mRNA differential display techniques and suppression subtractive hybridization are available for various aquatic organisms. In addition, the mushrooming of next-generation sequencing and the publication of genomes stimulated the transcriptome profiling analyses. As a powerful technology for obtaining massive amounts of data (Mohd-Shamsudinet al., 2013), transcriptome could offer the potential to explore the molecular mechanisms of heterosis by obtaining abundant differential expressed genes(DEGs) between parents and hybrids (Zhaoet al., 2019).Genome-wide changes in gene expression have been documented in yellow catfish (Zhanget al., 2019), black sea bream (Chenet al., 2020), sea cucumber (Wanget al.,2018b), and pearl oyster (Yanget al., 2018), while DEGs have been used to explain trait differences exhibited by hybrids.
The Pacific oysterCrassostrea gigasis one of the most economically important aquaculture species with high productivity and broad environmental tolerance. It has been introduced into many countries for aquaculture. However,C. gigassuffered mass mortality in both natural and cultured populations (Burdonet al., 2014), which has increased dramatically since 2008 and is considered to be resulted from the intricate interactions between living environment,opportunistic pathogens and oysters. Therefore, numerous studies on the oyster massive mortality during summer period have been conducted (Solomieuet al., 2015), and selective breeding has effectively reduced the mortality of oysters in the environment infected withOstreid herpesvirus 1(OsHV-1) (Garciaet al., 2011; Agnewet al., 2020).Nevertheless, hybridization is a more convenient and effective approach to improve the survival rate by crossing geographically isolated populations, selected lines and different species of oysters. For instance, hybridC. angulata×C. gigasexhibited higher cumulative survival rate under acute heat stress than their parental strains (Jianget al.,2022), crosses among three strains ofC. gigasresulted in the hybrids with better survival trait in natural environment(Konget al., 2017).
In our previous study, the hybrid oysters between female orange-shelled line and male ‘Haida No. 1’ line were produced, which have been demonstrated to exhibit heterosis in survival, stress resistance and growth (Menget al., 2021),especially the high heterosis in survival at planktonic larval stage (Lianget al., 2022). Notably, oysters carrya very high genetic load (at least 12 – 14 lethal genes per individual), and its mortality due to lethal recessive genes occurs primarily in larvae stage (Yin and Hedgecock, 2021). Crossbreeding could improve genetic heterozygosity and weaken the effect of recessive lethal genes, thereby increasing the survival of hybrids and generating heterosis (Whitlocket al., 2000). Therefore, the hybrid larvae could be used as a good model for oyster heterosis research, whose molecular basis remains unclear. In this study, the crossbreeding was carried out between two lines ofC. gigas, and heterosis of hybrid combination in larval survival rate was analyzed. The transcriptome analysis was conducted to reveal the molecular mechanism underlying the high ascendancy of survival rate in intraspecific hybrids. These results may provide clues to understand how hybrid oysters have a superior vitality than parental groups in a suitable breeding environment, which also increases our understanding of heterosis in oyster.
2 Materials and Methods
2.1 Experimental Animals
The Pacific oystersC. gigasused in this study were an inbred orange-shelled line (O, 10th generation) and a selected strain ‘Haida No. 1’ (H, 13th generation). The H strain with fast growth was successively selected using mass selection (Liet al., 2011), and the O line is a stable genetic line established by using the orange shell mutation individuals found in the self-bred offspring of purplish black shellC. gigas, which has poor adaptability to environmental stress and slow growth rate (Hanet al., 2019).
In April 2021, one-year-old oysters of H and O lines were collected from Sanggou Bay in Rongcheng (37.1˚N, 122.5˚E,Shandong, China) to conduct a crossbreeding experiment.Eggs and sperm were collected from females and males by dissection. Then, eggs from one female of O and sperm from one male of H were divided into two parts equally. Three families were established by the hybridization trial with pair mating combinations: H ♀ × H(HH), O ♀ × H(OH),and O ♀ × O(OO), and each family was divided into three incubators as three biological repeats. The larval rearing procedure was conducted as previous study of Liet al.(2011).
2.2 Measurement and Sampling
The larval survival rates of each family on days 1, 4, 9,13, 17, 21 and 25 after fertilization were calculated according to Konget al. (2017), and were presented as the mean± SD (n= 3) in table. Statistical significance was analyzed with one-way analysis of variance followed by multiple comparison Tukey test using SPSS 25.0 software, and the statistical significancewas considered ifP< 0.05. Also, the best-parent heterosis (BPH) and mid-parent heterosis (MPH)were calculated with the following formulas (Guoet al.,2017):
whereBP= the performance of best parental family, and
whereF1= the mean performance of hybrid family;MP=the mean performance of parental families.
At day 25 after fertilization, the eyed-pediveliger larvae of each family were sampled, respectively, and then a total of 9 samples were flashily transferred into RNAlater™stabilization solution and stored at −20℃.
2.3 RNA Extraction, Library Construction and Sequencing
Approximately 50 mg of each sample was used for extracting total RNA with TRIzol reagent (Invitrogen) according to the manufacturer’s instruction. The quality of RNA was confirmed by 1% agarose-gel electrophoresis. RNA purity and integrity were assessed using NanoPhotometer®spectrophotometer (IMPLEN) and RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies),respectively.
One μg RNA per sample was employed for further library construction. Following the instructions of manufacturer, 9 cDNA libraries were produced using NEBNext®UltraTMRNA Library Prep Kit for Illumina® (NEB), and index sequences were added to distinguish sample sequence.Firstly, the mRNA with PolyA tail was purified by Oligo(dT) magnetic beads from total RNA and then was broken into short fragments using NEB Fragmentation Buffer (5×).Subsequently, in the M-MuLV Reverse Transcriptase (RNase H-) system, the first strand cDNA was synthesized using mRNA fragments as templates and using random hexamer as primer. After degrading RNAviaRNase H, DNA Polymerase I system and dNTPs were added to synthesize the second strand cDNA. After purification, end-repair and Atailing of cDNA fragments, NEBNext Adaptor with hairpin loop structure was ligated. cDNA fragments ranging from 250 bp to 300 bp were selected by AMPure XP system and enriched by PCR, which were subsequently purified again. Lastly, the quality of cDNA library was assessed by the Agilent Bioanalyzer 2100 system. After clustering the index-coded samples on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia),the library preparations were sequenced by the Illumina Novaseq platform to generate 150 bp paired-end reads.
2.4 Data Analyses
The image data of sequence fragments were converted into raw reads in fastq format by CASAVA base recognition. Raw reads after quality control to remove reads containing adapter, ploy-N and low-quality reads became clean reads with high quality. Then, Q20, Q30 and GC content were calculated for clean reads. TheC. gigasreference genome and gene model annotation files were acquired from the genome website (ftp://ftp.ncbi.nlm.nih.gov/genomes/Crassostrea_gigas/). HISAT2 (v2.0.5) was used to construct the index of reference genome and align paired-end clean reads to the reference genome (Zhanget al., 2017), which was selected as the mapping tool on account of generating a database of splice junctions based on the gene model annotation file. The number of reads mapped to each gene was counted by Feature Counts (v1.5.0-p3) (Liaoet al.,2014).
2.5 Differential Expression Analysis
The correlation of samples was analyzed with RStudio v4.1.1 software. Differentially expressed genes (DEGs) of three families (three biological replicates per family) were identified using DESeq2 (1.16.1) with |log2(Fold change)|> 1 and a false discovery rate (FDR) < 0.05. R package clusterProfiler (v4.0.5) was used to process the GO and KEGG analyses, setting parameters as ‘pvalueCutoff = 0.05, pAdjustMethod = BH’ and ‘pvalueCutoff = 0.05’, respectively.For ease of description, the comparison of hybrid group OH with maternal group OO was defined as OH-M, and the comparison with paternal group HH is defined as OH-P.
2.6 Quantitative Real-Time PCR(qRT-PCR) Validation
To validate the results of RNA-seq, eight DEGs were selected for qRT-PCR analysis, and the gene-specific primers were designed using Primer Premier 5.0 (Table 1). 24 samples, eight biological replicates at per family, were selected for RNA extraction. After the assessment of integrity and purity, total RNA was reverse-transcribed into cDNA. In addition, elongation factor I-α (EFI-α) was used as an internal control to normalize the expression of target genes.qPCR was performed by ChamQ SYBR Color qPCR Master Mix (Vazyme). 2−ΔΔCtmethod was used to calculate the expression levels of target genes (Pfafflet al., 2002), and the data were analyzed usingt-test by SPSS 25.0 Statistics Software (IBM). The statistical significancewas considered at two-tailedP-value less than 0.05.

Table 1 Primers of genes selected for real-time qPCR
3 Results
3.1 Comparison of the Survival Rate of Three Families
Around 5 days postfertilization, the umbo larvae of each family have the highest mortality (Table 2). At day 25, the survival rates of three families were significantly different(P< 0.05), and the survival rate of hybrid family (27.67%)was higher than those of parental families (16.36% and 11.60%). Meanwhile, all the MPH and BPH of hybrid cross were positive and at a high level, which are in the ranges of 39.27% to 97.89% and 28.79% to 71.75, respectively(Table 2).

Table 2 Comparison of survival rate and heterosis among different families at larval stage of Crassostrea gigas
3.2 Transcriptome Profiling and Mapping
In total, an average of 48.16 million 150-bp paired-end raw reads per sequencing sample (42.83 – 54.89 million raw reads) were acquired. Then 42.50 – 51.57 million clean reads were filtered from each specimen, with Q20 (%)varying from 97.32% to 97.91%. The bases of clean reads exceeded 6.3 Gb, while 81.15% – 84.46% of the reads were aligned to the referenceC. gigasgenome, as shown in Table 3. The raw reads have been submitted to the SRA database of NCBI with the accession number PRJNA90-0427.

Table 3 Transcriptome mapping statistics
3.3 Analysis of Differentially Expressed Genes
DEGs between the hybrid group and parental groups were investigated by performing pairwise comparisons. Differences in gene expression (284 up-regulated DEGs and 141 down-regulated DEGs) of OH-P were identified. Meanwhile, 512 DEGs were observed in OH-M, including 410 up-regulated genes and 102 down-regulated genes. We further investigated the DEGs among two parental families(HHvs. OO), of which 685 genes were up-regulated and 481 genes were down-regulated (Fig.1 and Table 4). Among the DEGs, the number of up-regulated genes was higher than that of down-regulated genes.

Fig.1 Volcano plot based on different comparisons. A, OH vs. HH; b, OH vs. OO; c, HH vs. OO.

Table 4 Numbers of differentially expressed genes between inbred and hybrid families
3.4 Functional Analysis of DEGs
GO enrichment analysis was performed to identify the biological functions of DEGs between the hybrid group and parental groups, which may be involved in survival heterosis. The DEGs of OH-P were found mainly enriched in‘integrin activation’, ‘cellular extravasation’, ‘regulation of blood coagulation’ and so on (Fig.2), and the DEGs of OHM were significantly enriched in 47 GO terms (Fig.3). In order to further understand the metabolic processes and signal transduction pathways, KEGG enrichment analysis was implemented. The results showed that DEGs of OH-P were significantly enriched in the following pathways: ‘C-type lectin receptor signaling pathway’, ‘adherens junction’, ‘microRNAs in cancer’, ‘rap1 signaling pathway’, ‘endocrine resistance’, ‘human cytomegalovirus infection’ and ‘thyroid hormone signaling pathway’, while only pathway ‘inflammatory mediator regulation of TRP channels’ was enriched in OH-P DEGs (Table 5).

Fig.2 Scatter plot of GO enriched results of DEGs between OH and HH.
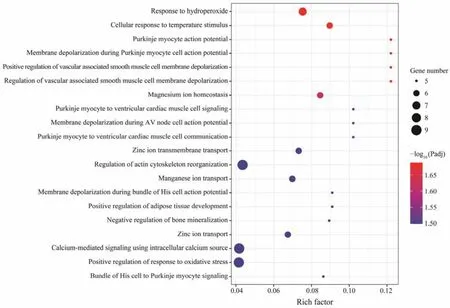
Fig.3 Scatter plot of top 20 GO enriched results of DEGs between OH and OO.

Table 5 Significantly enriched KEGG pathways (P < 0.05) of the DEGs among different comparisons
The results of GO analysis showed that 1166 DEGs between parental groups were enriched in 37 terms, which are related to immune response, synthesis of glycogen and lipids (Fig.4). These 1166 DEGs were also included in the following pathways: ‘arachidonic acid metabolism’, ‘steroid hormone biosynthesis’, ‘glutathione metabolism’, ‘retinol metabolism’ and ‘inflammatory mediator regulation of TRP channels’ (Table 5).
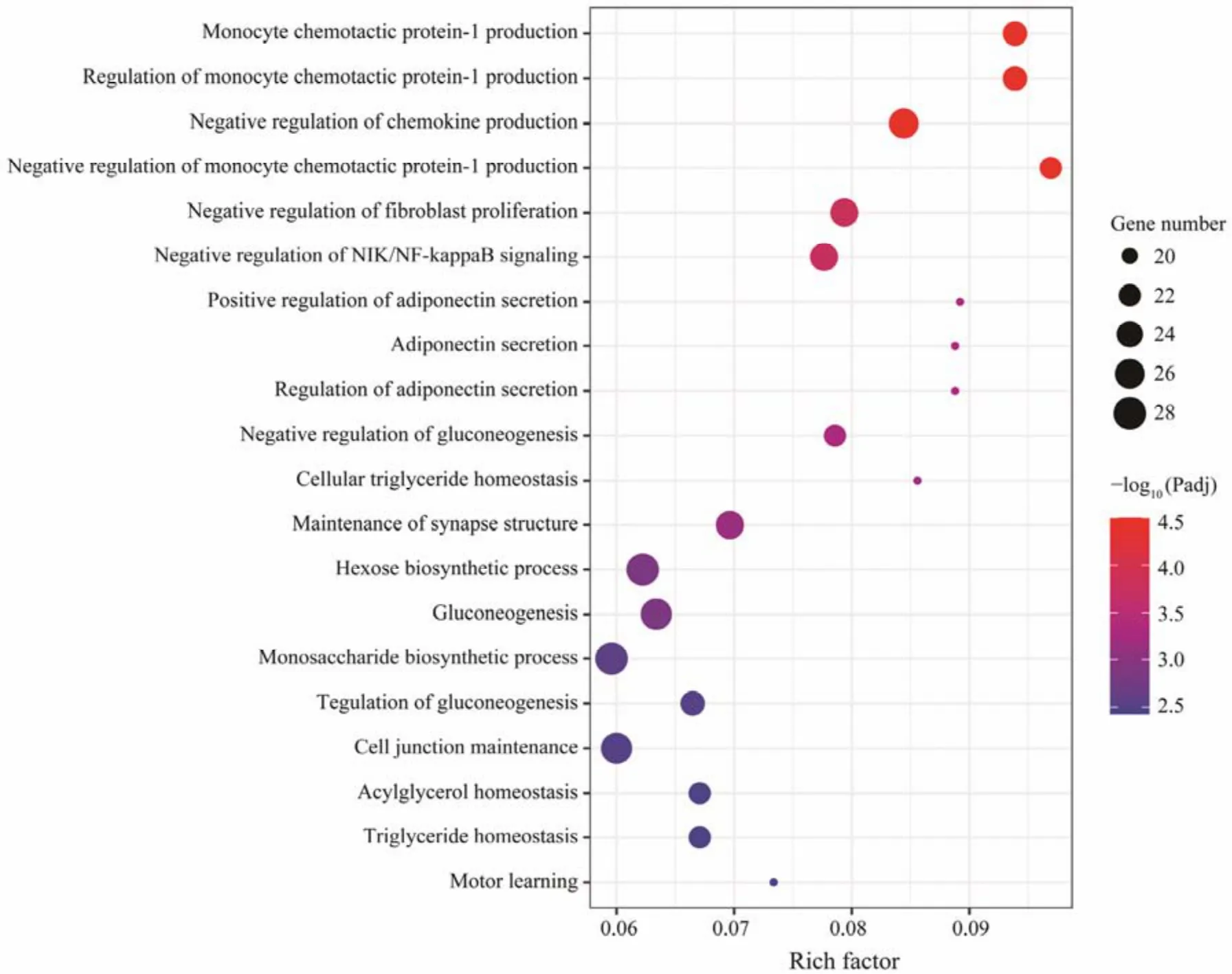
Fig.4 Scatter plot of top 20 GO enriched results of DEGs between HH and OO.
3.5 Validation of Differentially Expressed Genes Using qPCR
The qPCR validation of eight DEGs between different families were performed to confirm the accuracy of RNAseq results. Although the fold changes of DEGs calculated by qPCR were not completely consistent with that calculated by RNA-seq analysis, the direction of genetic expression change detected by qPCR was similar to those from RNA-seq (Fig.5), which confirms the accuracy and reliability of the RNA-seq method.

Fig.5 Validation of differentially expressed genes by real-time PCR. Error bars represent standard errors from biological replicates, and different letters indicate a significant differences at P < 0.05.
4 Discussion
Survival is regulated by a variety of complex biological processes and is implicated in multiple cellular processes.Here, the heterosis in survival ofC. gigashybrid combination was observed. After 25 days of culture, OH exhibited significant survival advantages over HH and OO. Such hybrid oysters with stronger environmental adaptability have also been observed in other studies:C. sikamea♀ ×C. gi-gas(Zhanget al., 2022b), northernC. ariakensis× southernC. ariakensis(Qinet al., 2022),C. gigas×C. angulata(Tanet al., 2020; Jianget al., 2022). However, there is little evidence to elucidate the molecular mechanism of heterosis in oysters, which is compounded by genome interactions and complex modifications at epigenetic and regulatory network levels (Sekinoet al., 2019). Recently, benefiting from the development of bioinformatics and highthroughput sequencing technologies, substantial efforts have been made in unveiling heterosis of bivalve shellfish at the molecular level by transcriptome sequencing (Geet al.,2008). For instance, transcriptome analysis of abalone revealed that more non-additive expressed genes and alternative splicing events may contribute to thermal heterosis of hybrid (Xiaoet al., 2021). Yanget al. (2018) found several growth-related genes in the DEGs between hybrids and their parents. In this study, we found several immune-related genes in the DEGs between hybrid OH family and parental families, which might contribute to the high heterosis in survival in hybrid ofC. gigas.
Transcriptome comparative analysis between OH group and OO group was performed. A total of 368 genes were identified as significantly differentially expressed, and several DEGs were related to immunoreaction by functional analysis. The perlucin-like protein is a regulating C-type lectin that interacts with the complement pathway and bacterial surface ligands, which functions in immune recognition (Moreiraet al., 2014). InMytilus galloprovincialis, perlucin-like gene was up-regulated afterVibrioinfection, indicating the possible role of this gene in innate immune response reactions in bivalves. In this study, the up-regulated perlucin-like gene may enhance the resistance of hybrid oyster. CD209 antigen is also a member of C-type lectin that plays an essential role in cell adhesion and pathogen recognition. In mammals, it is implicated as mediators of viral pathogenesis (Amraeiet al., 2021). InSalmo salar,CD209 antigen-like protein was involved in innate immunity (Sunet al., 2022). Additionally, a gene with high homology to the CD209 antigen-like protein (SsCTL4) was also identified in the black rockfish and demonstrated to promote bactericidal activity as a pattern recognition receptor (Duet al., 2018). Therefore, the higher expression level of CD209 antigen-like protein in OH family than parental families may be one reason for oyster heterosis.NFX1-type zinc finger-containing protein 1 (ZNFX1), an interferon (IFN)-stimulated and mitochondrial-localized dsRNA sensor, could specifically restrict the RNA virus replication by inducing the expression of IFN and ISG expression (Wanget al., 2019). In the process of viral defense, the up-regulation of ZNFX1 in OH oyster may enable the body to produce less inflammatory response through a post-transcriptional regulatory program.
Based on the integrated analysis of DEGs between OH and HH, C-type lectin receptor signaling pathway including caspase-6 and acan gene suggests its vital role in heterosis in survival of OH. Caspases could regulate immune responses, cell death, and homeostasis. The previous results suggested that caspase-3 exhibited caspase activity and could activate a variety of apoptosis-related substrates inC. gigas(Xuet al., 2016). Moreover, as an important executioner caspase, caspase-6 can activate caspase-3, leading to apoptosis (Ummanniet al., 2010). Thus, we speculated that the up-regulated caspase-6 in hybrid oysters may reflect an elevated level of cell apoptosis which can enhance the immune response and contribute to the lower mortality. Aggrecan,the proteoglycan family member, is the product of the aggrecan core protein gene (acan), which is the major component of extracellular matrix. Aggrecan relies on its glycosaminoglycans (GAGs) to combine with different protein ligands for biological functions, and it has been implicated in host defense, structural tissue organization, tissue coagulation and basement membrane integrity (Pomin and Mulloy, 2018). Some soluble proteoglycans may resist viruses and bacteria through their ectodomains acting as a protective agent. InM. galloprovincialisfollowingVibriochallenges, aggrecan plays a possible role in counteracting withVibriosurface ligands acting as soluble forms. Also, the presence ofVibrioincreased the expression level of aggrecan inBathymodiolus azoricus, which suggested that aggrecan is involved in innate immune response reactions of bivalves(Martinset al., 2014). In view of these functions, up-regulated acan in OH may contribute to the enhancement of host immune defense.
The DEGs between two inbred families (HH and OO)presented genetic differences in parental breeders, which is concerned with biological features in the two oyster lines.The cytochrome P450 (CYP450) is a family of hemoglobin-coupled monooxygenases. And a CYP450 gene cluster has been reported in Zebra Finch, showing the vital role of controlling red carotenoid coloration (Mundyet al., 2016).In QN orange scallops, CYP450 might affect orange coloration by the accumulation of melanin (Song and Wang,2019). Liet al. (2021) suggested the diverse differential expressions of CYP450 gene family between orange and black shell phenotype ofC. gigasmay regulate shell pigmentation. In this study, four CYP450 genes were differentially expressed among two parental groups, which may have been related to the orange shell of maternal families(OO). Additionally, we found several immune-related genes in the DEGs, which could be the reason for low survival rate of OO group. The F-type lectin (fucolectin) family includes fucose-binding proteins involved in innate immunity,which play a pathogen recognition and binding role in aquatic animals (Shaoet al., 2018). In pearl oyster, fucolectin-1 exhibited complex expression pattern changes under hightemperature induction (Zhanget al., 2022a). Wanget al.(2018a) identified a fucolectin fromApostichopus japonicus, indicating it was involved in the innate immune response of sea cucumber as a receptor with broad spectrum of microbial recognition. The down-regulated fucolectin genes (fucolectin-like, fucolectin-1 and fucolectin-3) might result in weak innate immune defense against bacterial infection of OO. The perlucin-like gene mentioned above also expressed differentially between two parental families(down-regulated in OO), which may be associated with the lower survival rate of OO oyster. Certainly, the precise roles of these genes in oyster need to be further investigated.
5 Conclusions
The heterosis in survival of larval stage in OH group was manifested in this study by comparing survival rate among the oysters from HH, OO and OH families. And the transcriptome analysis of hybrid oyster HO and parental families (HH and OO) was performed using RNA sequencing.We found that DEGs in the hybrid compared with its parents might be associated with survival heterosis. The findings of these DEGs might provide further insight into understanding the molecular mechanisms of heterosis in oysters.
Acknowledgements
This research was supported by the grants from the China Agriculture Research System Project (No. CARS-49),and the Earmarked Fund for Agriculture Seed Improvement Project of Shandong Province (No. 2020LZGC016).
 Journal of Ocean University of China2024年1期
Journal of Ocean University of China2024年1期
- Journal of Ocean University of China的其它文章
- Using Natural Radionuclides to Trace Sources of Suspended Particles in the Lower Reaches of the Yellow River
- Eutrophication of Jiangsu Coastal Water and Its Role in the Formation of Green Tide
- Evaluation of the Shallow Gas Hydrate Production Based on the Radial Drilling-Heat Injection-Back Fill Method
- Microstructure Characterization of Bubbles in Gassy Soil Based on the Fractal Theory
- Morphological and Sulfur-Isotopic Characteristics of Pyrites in the Deep Sediments from Xisha Trough, South China Sea
- Deformation Characteristics of Hydrate-Bearing Sediments