
喹啉酮类BRD4 抑制剂的3D-QSAR 研究
2024-03-11 08:30:42刘亚平张淑平
广州化学 2024年1期
刘亚平,程 平,张淑平
(上海理工大学 材料与化学学院,上海 200093)
含溴域蛋白4(bromodomain-containing protein 4,BRD4)[1]参与调控细胞周期、增殖和凋亡等活动。开发BRD4 抑制剂被认为是一种治疗癌症的有前景的策略[2]。近年来,计算机辅助药物设计逐渐成为开发和改造药物的重要方法之一,在高效开发新型BRD4 抑制剂方面具有广泛的应用前景。
尽管多种BRD4 抑制剂用于临床阶段的研究,但临床试验过程中所表现出的疗效差和耐药性等问题不容忽视,仍需研发出更多高活性的BRD4 抑制剂。传统的药物设计方法通常周期长达10 年,成本高达26 亿美元,无法满足临床药物需求[3]。
近年来,3D-QSAR 研究以更经济高效的方式加快药物研发效率而得到广泛关注[4]。为了设计出高活性的喹啉酮类小分子p53-MDM2 结合抑制剂,尤等人建立了具有预测能力的3D-QSAR 模型[5]。Tong等利用一系列4,5-二氢-[1,2,4]三唑并[4,3-f]蝶啶类BRD4 抑制剂建立QSAR 模型,并成功设计出4 个新BRD4 抑制剂[6]。此外,基于CoMFA 和CoMSIA两种不同的分析方法,Yu 等人使用一系列四氢喹喔啉类BRD4 抑制剂建立了3D-QSAR 模型,并获得5 个高活性的候选化合物,为新型BRD4 抑制剂的合成和设计提供必不可少的理论依据[7]。很多研究者对各类BRD4 抑制剂进行QSAR 建模,并在开发新型BRD4 抑制剂方面取得了许多成果,但是关于喹啉酮类BRD4 抑制剂的3D-QSAR 相关研究还比较匮乏。
在现有CADD 技术的启发下,本文利用已报道的一系列BRD4 抑制剂建立3D-QSAR 模型并开发了一系列新型BRD4 抑制剂。通过三维比较分子力场分析(comparative molecular field analysis,CoMFA )[8]和比较分子相似性指数分析(comparative molecular similarity index analysis,CoMSIA)[9]预测BRD4 抑制剂的结构与活性之间的关系。此外,为了评估新设计分子的药效,对其进行ADMET 性质和类药性评价。所有研究成果为更高效地设计新型BRD4 分子提供了理论参考。
1 实验
1.1 数据集构建和分子叠合
从文献中提取33 个生物活性数据已知的喹啉酮类化合物,选取其中的27 个化合物作为训练集来构建QSAR 模型,剩下的6 个化合物组成测试集来验证模型的预测能力。33 个化合物的结构和生物活性数据如表S1 所示[10]。
本研究使用SYBYL-X 2.0 软件绘制分子的化学结构并进行几何优化,然后使用最陡梯度下降法对这些结构进行能量最小化计算,从而获得更稳定的构象。原子电荷的计算和分子的能量最小化优化步骤是在Chaube 等人的工作基础上进行了改进,将最大迭代次数设置为10000 次[11]。随后,将活性最高的化合物30 作为模板分子,并选择一个合适的公共骨架进行分子叠合。选择的公共骨架和训练集分子叠合结果如图1 所示。
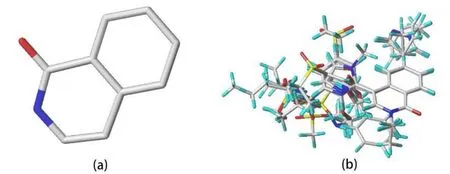
图1 BRD4 抑制剂的(a)公共骨架图和(b)28 个分子叠合图
1.2 CoMFA 和CoMSIA 模型构建和PLS 分析
为了探究抑制剂的生物活性与分子力场的关系,进行了CoMFA 和CoMSIA 模型的构建,模型构建过程中参考了李等人的方法和参数设置[12]。CoMFA 计算包括静电场(electrostaic,E)和立体场(steric,S)。而CoMSIA 计算除了静电场和立体场外,还有疏水场(hydrophobic,H)、氢键受体场(acceptor,A)和氢键供体场(donnor,D)。使用偏最小二乘(partial least-square,PLS)方法建立具有统计学意义的CoMFA 和CoMSIA 模型,将pIC50 值作为因变量,CoMFA 和CoMSIA 描述符作为自变量。在PLS 分析的交叉验证方法中,采用留一法(left-one-out,LOO)得到最优组成分值(optimal number of components,ONC)和交叉验证回归系数(cross-validation correlation coefficient,q2)。然后使用非交叉验证方法得到标准误差(standard error of estimation,SEE)、Fischer 检验值(Fischer test,F)和非交叉验证相关系数(square of the non-cross-validation correlation coefficient,r2),这些参数用于评价模型的预测能力和稳定性。此外,为了进一步评估3D-QSAR 模型的预测能力,利用建好的模型来预测测试集中所有化合物的pIC50 值,并通过计算预测相关系数r2pred来评估模型的外部预测能力。
1.3 ADMET 药物动力学预测和类药性分析
ADMET(药物在人体内的吸收、分布、代谢、排泄和毒性)药物动力学方法是药物设计和筛选的重要方法。在药物开发早期,使用ADMET 药物动力学方法对候选化合物进行预测,可以有效提高药物研发和筛选成功率,减少资金的浪费。在本研究中 , 使 用 pkCSM[13]在 线 服 务 器(http://biosig.unimelb.edu.au/pkcsm/prediction)进行ADME 和毒性评估。此外,类药性评估也是识别潜在的候选药物的重要手段,广泛使用的SwissADME[14]工具(http://www.swissadme.ch)可以预测新设计分子的合成可行性和类药性。
2 结果与讨论
2.1 CoMFA 和CoMSIA 研究
经过PLS 分析得到的CoMFA 和CoMSIA 模型相关统计参数如表1。在CoMFA 模型中,立体场和静电场的贡献率分别为0.610 和0.390,经过交叉验证得到q2为0.926,ONC 为7。采用非交叉验证得到的r2为0.997,SEE 为0.078,F为907.047,以及经过计算得到r2pred为0.744。由于CoMSIA 计算中包含五种不同的立场,为了获得性能更优的CoMSIA 模型,选取了不同的立场进行CoMSIA 建模,各个模型的PLS 结果如表2 所示,一般情况下,q2>0.5,r2pred>0.7,说明该模型具有良好的鲁棒性和预测能力,构建的所有模型的q2和r2值均接近1,说明这些模型均具有较好的稳定性和预测能力。其中SEHA 组合的CoMSIA 模型的q2和r2值分别为0.939 和0.991,具有更突出的预测能力。因此,选择CoMSIA-SEHA 模型进行后续的分析和使用。在CoMFA 模型和CoMSIA-SEHA 模型中,训练集化合物的实验值和预测pIC50 值的散点图如图2 所示。从图中可以观察到,训练集(黑点)和测试集(红点)都均匀散布在直线y=x周围,进一步说明建立的模型具有良好的预测能力和拟合能力。
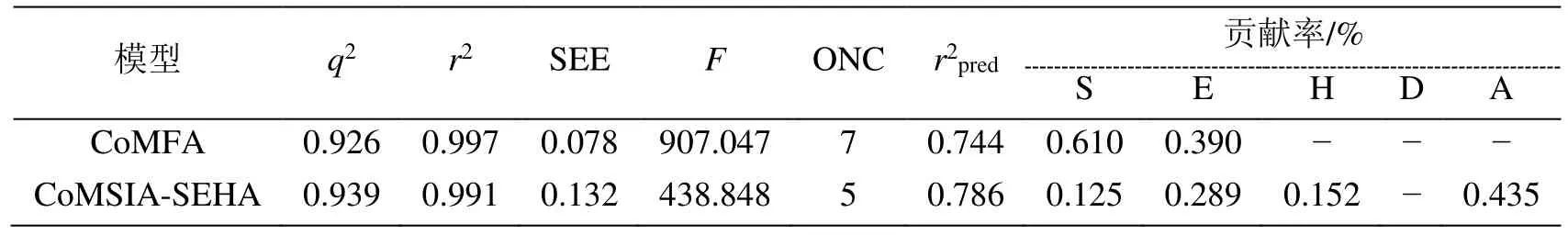
表1 CoMFA 和CoMSIA 模型统计分析总结

表2 不同立场组合的CoMSIA 模型PLS 结果

图2 CoMFA 模型和CoMSIA 模型的实验pIC50 值与预测pIC50 值散点图
2.2 3D-QSAR 等势图分析
结合活性最好的化合物30,对CoMFA 和CoMSIA 等势图进行合理分析,发现能提高化合物活性的取代基,得到更多分子改造的重要信息,进而为设计出理论活性更好的化合物提供理论指导。
CoMFA 等势图的立体场和静电场如图3 所示,在立体场中,绿色区域(贡献度为80%)表示增大基的空间位阻有利于增加化合物活性,而黄色区域(贡献度为20%)表示减小基团会增加化合物活性。在图3a 中,A 环周围存在大面积的黄色区域,表明此区域内合理减少基团的空间位阻,能促进化合物活性增加。化合物13(pIC50=6.149)比化合物14(pIC50=5.524)的活性高,也许是因为前者的甲基空间位阻明显小于后者的苯基空间位阻。在静电场中,蓝色区域(贡献度为80%)表示增加正电性基团有利于增加化合物活性,而红色区域(贡献度为20%)表示增加负电性基团有利于增加化合物活性。从图3b 中可以看到,A 环由大量蓝色色块包围,表明该区域加入吸电子基团有助于提高化合物生物活性,这也解释了含有给电子基团-NH2的化合物 32(pIC50=7.310)的活性远小于化合物 33(pIC50=8.046)。
图3 基于3D-QSAR 的结构与活性关系的主要特征
CoMSIA 等势图如图4 所示,图4a 和图4b 表示出的立场信息与图3 类似。图4c 表示疏水场,黄色色块表示增加疏水基团有利于增加活性,而白色表示增加亲水性基团更有利于提高化合物的生物活性。图中A 环的第6 位置有白色色块,表面该位置降低疏水基团可有效增加化合物活性,化合物30(pIC50=8.523)在该区域的基团多为亲水性烃基,而化合物32(pIC50=7.310)在该位置的基团是疏水性胺基,所以前者的生物活性明显大于后者。图4d表示氢键受体场,明显的红色区域表明减少氢键受体有利于增加抑制剂的生物活性,含有氮和氧元素的A 环第3 位置被大块红色色块包裹,该现象可以解释为何化合物11(pIC50=5.469)的活性值小于化合物12(pIC50=6.292)。
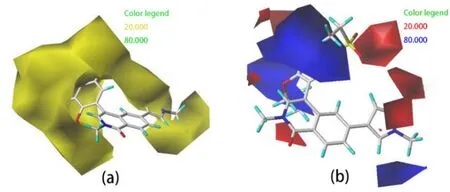
图4 模板化合物30 的CoMFA 等势图:a. 立体场;b. 静电场

图5 模板化合物30 的CoMSIA 等势图:a. 立体场;b. 静电场;c. 疏水场;d. 氢键受体场
2.3 新BRD4 抑制剂分子设计
根据等势图的分析结果,最终设计出7 个新化合物,这些新化合物的pIC50 值均大于活性最好的化合物30,其具体结构和预测值如表3 所示。
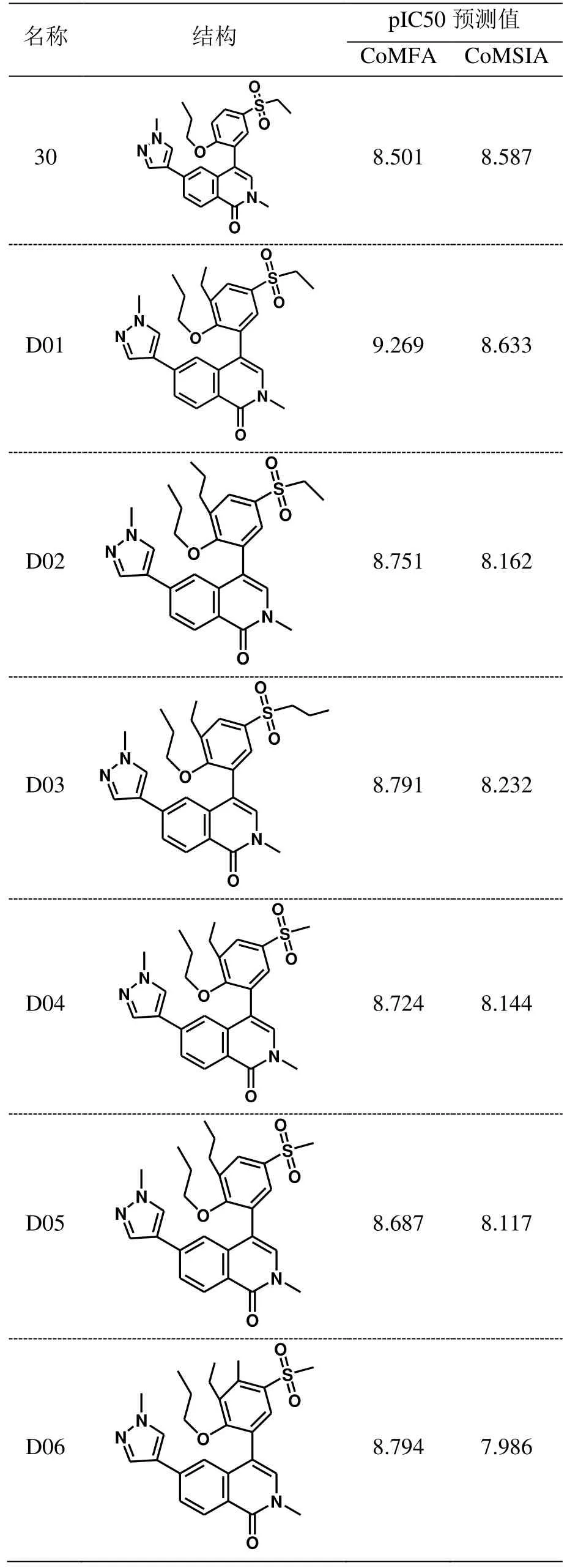
表3 新设计化合物的结构及预测活性
2.4 ADMET 属性预测和类药性分析
为了提高药物研发的成功率,减少药物毒性和副作用,并指导临床合理用药,在药物研发前期需要对药物进行药代动力学研究。对新设计的BRD4抑制剂进行ADMET 预测,同时进行了类药性研究,进一步分析这些抑制剂是否具备理想药物所表现出来的理化性质和结构特性。ADMET 性质预测结果如表4 所示,类药性预测如表5 所示。
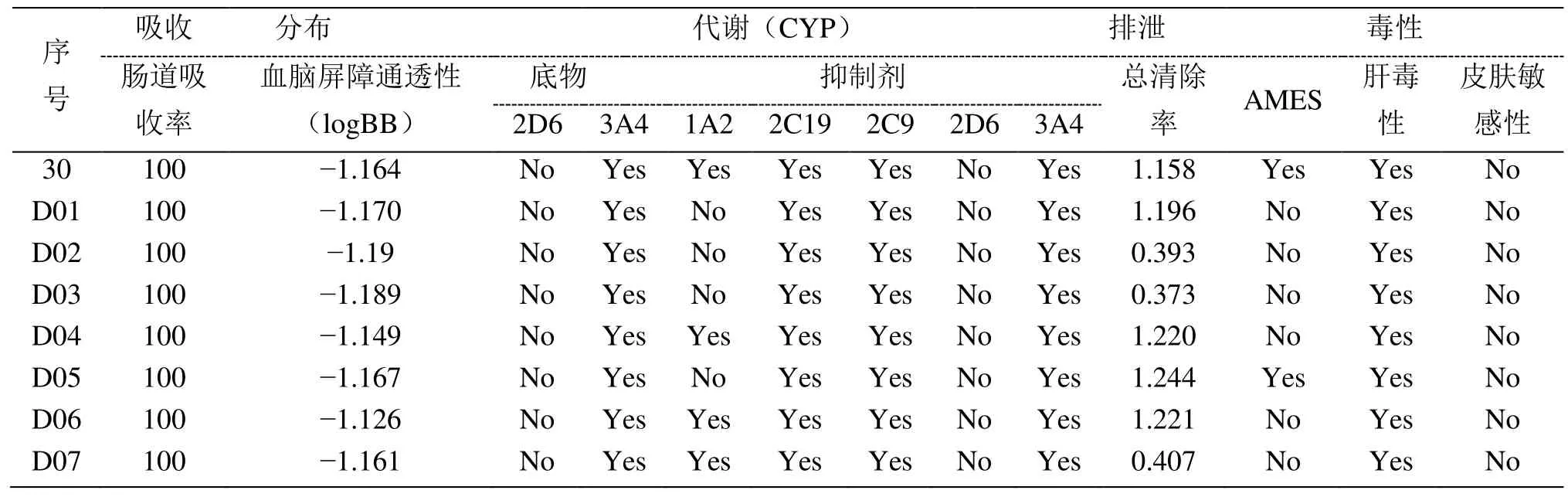
表4 新设计分子的ADMET 性质
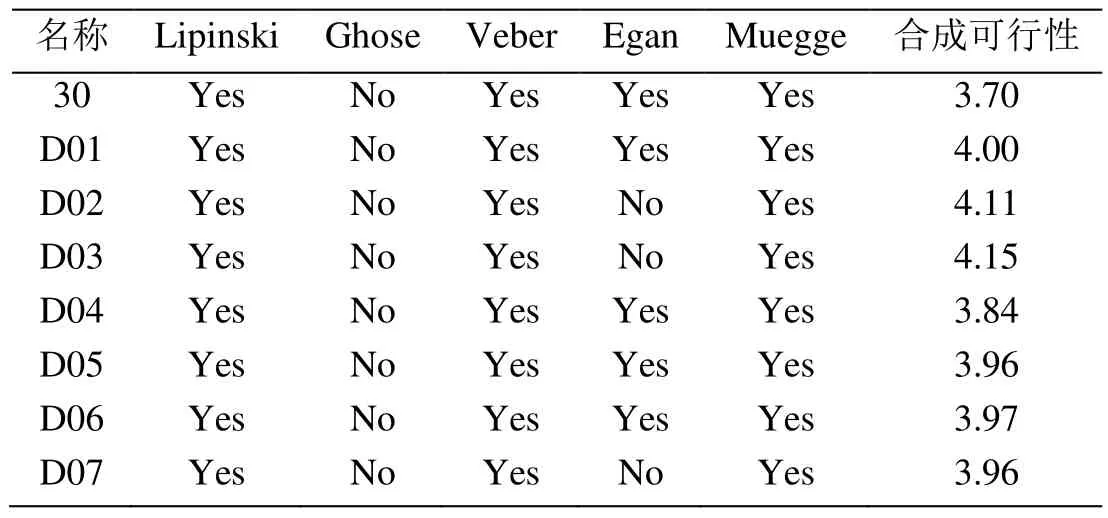
表5 新设计分子的药物相似性及其合成可达性
药物从作用部位进入体循环的过程中,需要考虑人体肠道吸收率,一般吸收率大于30%的分子则表现出良好的吸收性。表4 所示的7 个新分子的吸收率均大于30%,表现出出色的人体肠道吸收性。药物穿过血脑屏障的能力是提高药物利用效力、减少人体副作用的重要指标。通常情况下,logBB<-1表示该化合物穿透血脑屏障的能力较差。由表4 可知,7 个新分子均不易透过血脑屏障,所以具有很低的神经毒性。细胞色素P450 是存在于人体肝脏内的一种重要解毒酶,能影响药物的代谢,所以评估化合物抑制细胞色素P450 的能力至关重要。参与药物代谢的两个主要异构体是 CYP2D6 和CYP3A4,由表4 可知,7 个新分子均不是CYP2D6的底物和抑制剂,但属于CYP3A4的底物和抑制剂,所以可以被CYP3A4 正常代谢。药物清除率和肝脏清除率和肾脏清除率两者有关,表中的所有新分子均不容易被肝脏和肾脏联合清除。预测药物对人体的毒性也是提高药物成药性的重要步骤。所有新设计分子均不会导致皮肤敏感现场产生,除了D07,其他新设计化合物均没有AMES 毒性。总之,以上预测结果表明7 个新分子具有较为良好的药代动力学特性。
表5 的类药性预测结果表明,7 个新分子均符合Lipinski、Veber 和Muegge 规则,部分化合物也满足Egan 规则,但所有化合物均不满足Ghose 规则,说明所有新设计化合物具有理想的类药性。此外,所有化合物的可合成性均在3 左右,表现出易于人工合成的特点。以上研究结果,为缩小药物筛选范围提供有利帮助。
3 结论
本研究采用CoMFA 和CoMSIA 方法对一系列BRD4 抑制剂进行3D-QSAR 研究。通过内外部验证,CoMFA 模型(q2=0.926,r2=0.997,r2pred=0.744)和CoMSIA 模型(q2=0.939,r2=0.991,r2pred=0.786)均表现出优异的统计结果。进一步对模型的等势图进行分析,为设计活性更强的新BRD4 抑制剂提供理论指导。此外,对新设计的7 个BRD4 抑制剂进行ADMET 和类药性预测,证明了7 个新设计化合物具有良好的药代动力学特性。该研究结果为开发更高效喹啉酮类BRD4 抑制剂提供理论参考。
猜你喜欢
基层中医药(2020年12期)2020-07-22 06:35:00
铜仁学院学报(2018年6期)2018-07-05 09:47:34
上海农业学报(2017年3期)2017-04-10 12:39:26
衡阳师范学院学报(2016年3期)2016-07-10 07:16:27
中国资源综合利用(2016年10期)2016-01-22 08:36:17
中国卫生标准管理(2015年25期)2016-01-14 09:29:27
电测与仪表(2015年10期)2015-04-09 11:48:04
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01 02:53:54
中国当代医药(2015年16期)2015-03-01 02:03:13
中国药理学通报(2014年2期)2014-05-09 08:22:39
