
Gene signatures to therapeutics: Assessing the potential of ivermectin against t(4;14) multiple myeloma
2024-03-08 00:11YangSongHaoJunZhangXiaSongJieGengHongYiLiLiZhongZhangBoYangXueChunLu
Yang Song,Hao-Jun Zhang,Xia Song,Jie Geng,Hong-Yi Li,Li-Zhong Zhang,Bo Yang,Xue-Chun Lu
Abstract BACKGROUND Multiple myeloma (MM) is a terminal differentiated B-cell tumor disease characterized by clonal proliferation of malignant plasma cells and excessive levels of monoclonal immunoglobulins in the bone marrow.The translocation,(t)(4;14),results in high-risk MM with limited treatment alternatives.Thus,there is an urgent need for identification and validation of potential treatments for this MM subtype.Microarray data and sequencing information from public databases could offer opportunities for the discovery of new diagnostic or therapeutic targets.AIM To elucidate the molecular basis and search for potential effective drugs of t(4;14) MM subtype by employing a comprehensive approach.METHODS The transcriptional signature of t(4;14) MM was sourced from the Gene Expression Omnibus.Two datasets,GSE16558 and GSE116294,which included 17 and 15 t(4;14) MM bone marrow samples,and five and four normal bone marrow samples,respectively.After the differentially expressed genes were identified,the Cytohubba tool was used to screen for hub genes.Then,the hub genes were analyzed using Gene Ontology and Kyoto Encyclopedia of Genes and Genomes analysis.Using the STRING database and Cytoscape,protein-protein interaction networks and core targets were identified.Potential small-molecule drugs were identified and validated using the Connectivity Map database and molecular docking analysis,respectively.RESULTS In this study,a total of 258 differentially expressed genes with enriched functions in cancer pathways,namely cytokine receptor interactions,nuclear factor (NF)-κB signaling pathway,lipid metabolism,atherosclerosis,and Hippo signaling pathway,were identified.Ten hub genes (cd45,vcam1,ccl3,cd56,app,cd48,btk,ccr2,cybb,and cxcl12) were identified.Nine drugs,including ivermectin,deforolimus,and isoliquiritigenin,were predicted by the Connectivity Map database to have potential therapeutic effects on t (4;14) MM.In molecular docking,ivermectin showed strong binding affinity to all 10 identified targets,especially cd45 and cybb.Ivermectin inhibited t(4;14) MM cell growth via the NF-κB pathway and induced MM cell apoptosis in vitro.Furthermore,ivermectin increased reactive oxygen species accumulation and altered the mitochondrial membrane potential in t(4;14) MM cells.CONCLUSION Collectively,the findings offer valuable molecular insights for biomarker validation and potential drug development in t(4;14) MM diagnosis and treatment,with ivermectin emerging as a potential therapeutic alternative.
Key Words: Multiple myeloma;Functional enrichment analysis;Molecular docking simulation;Gene expression profiling;Therapeutic target;Ivermectin
lNTRODUCTlON
Multiple myeloma (MM) represents a severe hematological malignancy,affecting 176404 individuals and resulting in117077 fatalities annually[1].MM is characterized by uncontrolled plasma cell proliferation in the bone marrow,leading to severe complications,such as bone and kidney damage,anemia,and hypercalcemia[2].Of particular concern is the t(4;14) subtype,comprising up to 15% of new MM cases,with notably low survival rates and strong resistance to existing therapies[3].Thus,effectively addressing this subtype remains an important medical challenge.Although the seminal research of Foltzetal[4] and Ashbyetal[5] have provided insights into the pivotal facets of t(4;14) MM (Supplementary Table 1),which have advanced our understanding of this malignancy,certain aspects remain elusive.
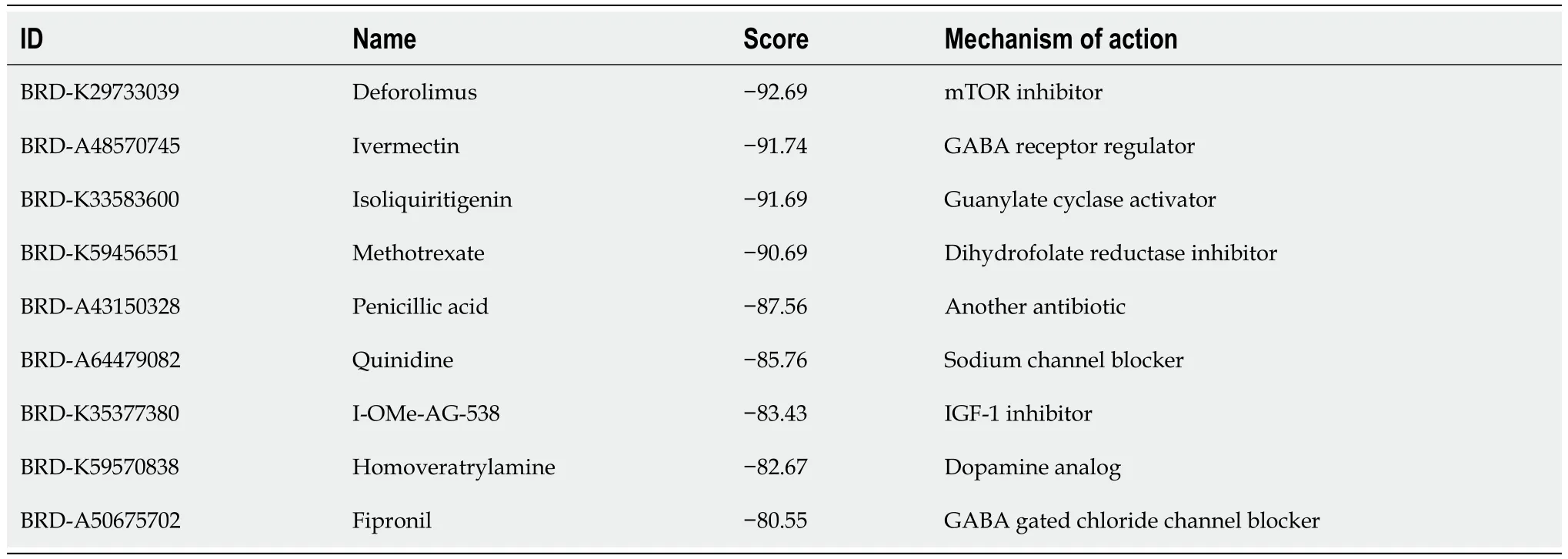
Table 1 Potential drugs identified using the Connectivity Map database
Translational medicine is increasingly considering drug repurposing as a potential strategy to develop efficient,safe,cost-effective,and readily available anticancer treatments[6].Advances in high-throughput sequencing technology have expanded biomedical and computational resources,facilitating a deeper understanding of cancer etiology and drugtarget interactions,thus enabling drug repurposing[7].Notably,the Connectivity Map (CMap) tool,housing a comprehensive dataset of 7000 microarrays from various cancer cells treated with 1309 molecular compounds,has been instrumental in identifying potential treatments for various cancers[8].Notably,Shietal[9] used CMap to identify eugenol as a potential treatment for triple-negative breast cancer,and Qiuetal[10] computationally identified small-molecule drugs with the potential to treat cervical cancer.
There is an urgent need for identification and validation of potential treatments for this MM subtype,therefore,we aim to elucidate the molecular basis and search for potential effective drugs by employing a comprehensive approach.We hope to contribute to advancing early MM diagnosis and tailoring its treatment.
MATERlALS AND METHODS
Differential expression analysis
The Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/gds) is a key public repository housing high-throughput gene expression data,microarray data,and gene chip information.For this study,we sourced the t(4;14) MM-related expression dataset from GEO.Our search parameters were tailored to t(4;14) MM across all fields,combined with filters for "Homo sapiens" as the organism and "dataset." Two datasets,GSE16558 and GSE116294,which included 17 and 15 t(4;14) MM bone marrow samples,and five and four normal bone marrow samples,respectively (Supplementary Table 2),were further considered for our analysis.To identify DEGs between the t(4;14) MM samples and normal samples,we used the "limma" package in R software [version 4.2.2,(http://www.R-project.org/)].Our selection criteria were aPvalue < 0.05 and absolute log2 fold change (FC) > 1.
PPI network analysis
To construct a functional PPI network for the identified DEGs,we used the STRING online database tool (https://stringdb.org/)[11].Our selected threshold required a credibility score of > 0.4 for inclusion within the network.The Cytoscape software [version 3.10.1,(https://cytoscape.org/)] was used to visually map the PPI,offering a clear understanding of protein interactions.
Hub gene mapping
Using Cytoscape,the molecular complex detection (MCODE) plugin was activated to highlight prominent clusters,abiding by criteria,such as an MCODE score > 6 and a node count > 4.The CytoHubba plugin[12] within Cytoscape,paired with the Maximal Clique Centrality (MCC) method,was used to filter and identify the top-ranking genes.A combination of results drawn from the MCODE,MCC,and degree scores led to the identification of 10 key hub genes.
Functional enrichment analysis
The enrichment analysis was performed using the “GSEApy” package within Python (v.3.11.4,[https://www.python.org]),using the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology.Subsequently,statistical significance was evaluated using the p-value,whereas the visualization of data was achieved using the “ggplot2” package in R.Gene Set Enrichment Analysis (GSEA) was performed using its dedicated software,sourcing gene sets from the Molecular Signature Database (http://software.broadinstitute.org/gsea/index.jsp).
Drug screening using CMap
The CMap database (https://clue.io)[13],a key tool in systems biology,was used to identify potential therapeutic candidates.Our criteria were rigorous,with a cutoff score of < -80.
Molecular docking
For our molecular docking investigations,3D structural models of the proteins encoded by target genes were retrieved from UniProt[14] and Protein Data Bank databases and augmented with AlphaFold data.Focusing on structures determinedviaX-ray diffraction with a co-crystal resolution < 2.5 Å,we downloaded the 2D structural model of ivermectin from PubChem[15] and converted it into a 3D structure using the LigPrep module in Maestro.AutoDockTools were used to preprocess the ligands.Optimization included water removal,hydrogenation,charge determination,and torsion centers and bond selection,yielding a pdbqt file suitable for docking studies.Macromolecular docking calculations were performed using Vina software [version 1.1.2,(http://vina.scripps.edu/)].To illustrate the postdocking ligand-receptor interactions,we used Ligplot+[16] for 2D mapping and PyMOL[17] for in-depth 3D analysis.
Reagents and antibodies
Ivermectin was purchased from Aladdin Biological Technology (Shanghai,China).The following primary antibodies were purchased from CST (Danvers,MA,United States): Anti-poly (ADP-ribose) polymerase (PARP) (9532;1:1000),anticleaved PARP (5625;1:1000),anti-cleaved caspase 3 (9661;1:500),anti-cleaved caspase 9 (20750;1:1000),and β-actin (3700;1:1000).The following primary antibodies were purchased from Abcam (Cambridge,United Kingdom): Anti-B-cell lymphoma 2 (Bcl2;ab 182858;1:1000),anti-Bcl-2-associated X protein (Bax;ab32503;1:1000),anti-NF-κB p65 (ab32536;1:1000),anti-NF-κB p65 (phospho S536) (ab86299;1:1000),anti-inhibitor of NF-κB (IκB) alpha (ab32518;1:1000),and anti-IκB alpha (phospho-S36) (ab 133462;1:1000).The following secondary antibodies were purchased from Beyotime Biotechnology (Shanghai,China): anti-rabbit IgG (A0208;1:5000) and anti-mouse IgG (A0216;1:5000).
Cell culture
NCI-H929,a t(4;14) MM cell line,was obtained from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences (Shanghai,China).The cells were cultured in RPMI-1640 medium (Gibco,Billings,MO,United States) with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (Gibco) at 37 °C with 5% CO2.Preliminary tests confirmed that the cell lines wereMycoplasma-free.
Cytotoxic activity assay
To assess cytotoxic activity,5000 cells were seeded per well into a 96-well plate and exposed to ivermectin concentrations ranging from 0-20 μmol/L for 24-48 h.After incubation,cell viability was evaluated using Cell Counting Kit-8 (CCK8) (DojinDo,Shanghai,China),which required an additional 2-h incubation with 10 μL of CCK8 solution at 37 °C.The absorbance of the reaction mixture was measured at 450 nm using a microplate reader (Thermo Fisher Scientific,California,United States).The inhibitory concentration 50% (IC50) of ivermectin for each cell type was determined from the viability data using GraphPad Prism v.10 and the following equation:
[(As-Ab)/(Ac-Ab)] × 100%,where “As,” “Ac,” and “Ab” are the absorbance values in the experimental,control,and blank wells,respectively.
Apoptosis assay
Cells were seeded into 6-well plates and treated with ivermectin for 24 h.After treatment,the cells were collected by centrifugation.They were then stained using the fluorescein isothiocyanate (FITC) Annexin V Apoptosis Detection Kit with 7-aminoactinomycin D (7-AAD; 640922;BioLegend,San Diego,CA,United States) by adding 5 µL of FITC conjugated with annexin V and 5 µL of 7-AAD solution.This was followed by 15 min incubation in the dark at 25 °C.Subsequently,the stained cells were analyzed using a flow cytometer (LSRFortessa;BD,Franklin Lakes,NJ,United States),and the data obtained were processed and interpreted using FlowJo software (v.10.4).
Reactive oxygen species assay
The cells were cultured in 6-well plates overnight.Fresh RPMI-1640 medium containing the indicated concentrations of ivermectin was added,and the cells were cultured for 24 h.A reactive oxygen species (ROS) assay kit (S0033S;Beyotime) was used to detect intracellular ROS levels using the following protocol: treated cells were incubated with 2’,7’-dichlorodihydrofluorescein diacetate for 20 min at 37 °C.Cells were washed thrice with serum-free medium,and images were acquired using a fluorescent inverted microscope (Olympus,Tokyo,Japan).
Mitochondrial membrane potential assay
Cells were first allowed to settle in 6-well plates,after which they were treated with ivermectin for 24 h.A JC-1 assay kit (M34152;Thermo Fisher Scientific) was used to evaluate the mitochondrial membrane potential within these cells.The assessment was performed in strict accordance with the manufacturer’s guidelines.Readings from this assessment were captured using a flow cytometer (LSRFortessa;BD).
Western blot analysis
After 48 h of ivermectin treatment,the cells were harvested and lysed using Cell Lysis Buffer (P0013;Beyotime),which included a protease inhibitor.The subsequent lysate was subjected to a 30-min ice bath before centrifugation at 12000 rpm for 20 min at 4 °C.Proteins were quantified using a BCA assay kit (P0010;Beyotime),separated by sodium dodecylsulfate polyacrylamide gel electrophoresis,and transferred onto polyvinylidene fluoride membranes.Subsequently,these membranes were blocked with 5% skim milk for 2 h and incubated with primary antibodies overnight at 4 °C.After thorough washing,the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at 25 °C.The proteins were visualized using an electrochemiluminescence kit (NCM Biotech,Suzhou,China).
Statistical analysis
All statistical evaluations were performed using both the R package and GraphPad Prism v.9.0 (GraphPad Software Inc.,San Diego,CA,United States).Student’st-test was used to compare the means between two groups.Comparisons across multiple groups were conducted using one-and two-way ANOVA.All results are presented as the mean ± SD.Statistical significance was set atPlevels < 0.05.
RESULTS
Identification of DEGs
We identified 1,100 DEGs (270 upregulated and 830 downregulated) and 1746 DEGs (808 upregulated and 938 downregulated) in the GSE16558 and GSE116294 datasets,respectively.These microarray findings have been presented using heat maps and volcano graphs,applying a threshold ofP< 0.05,logFC<1 for downregulated genes,and logFC>1 for upregulated genes (Supplementary Figure 1).
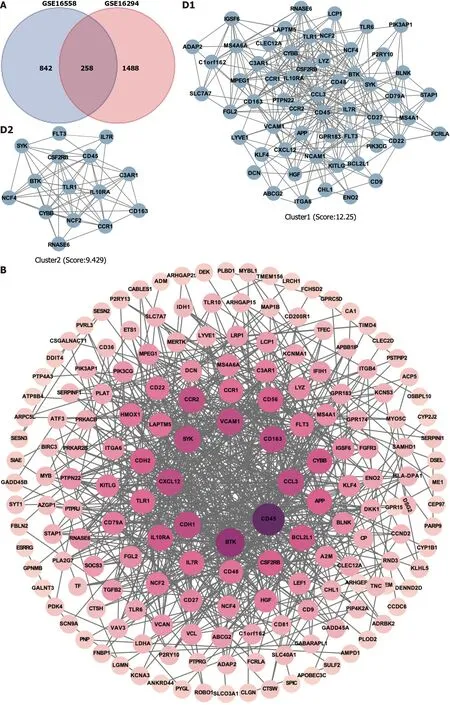
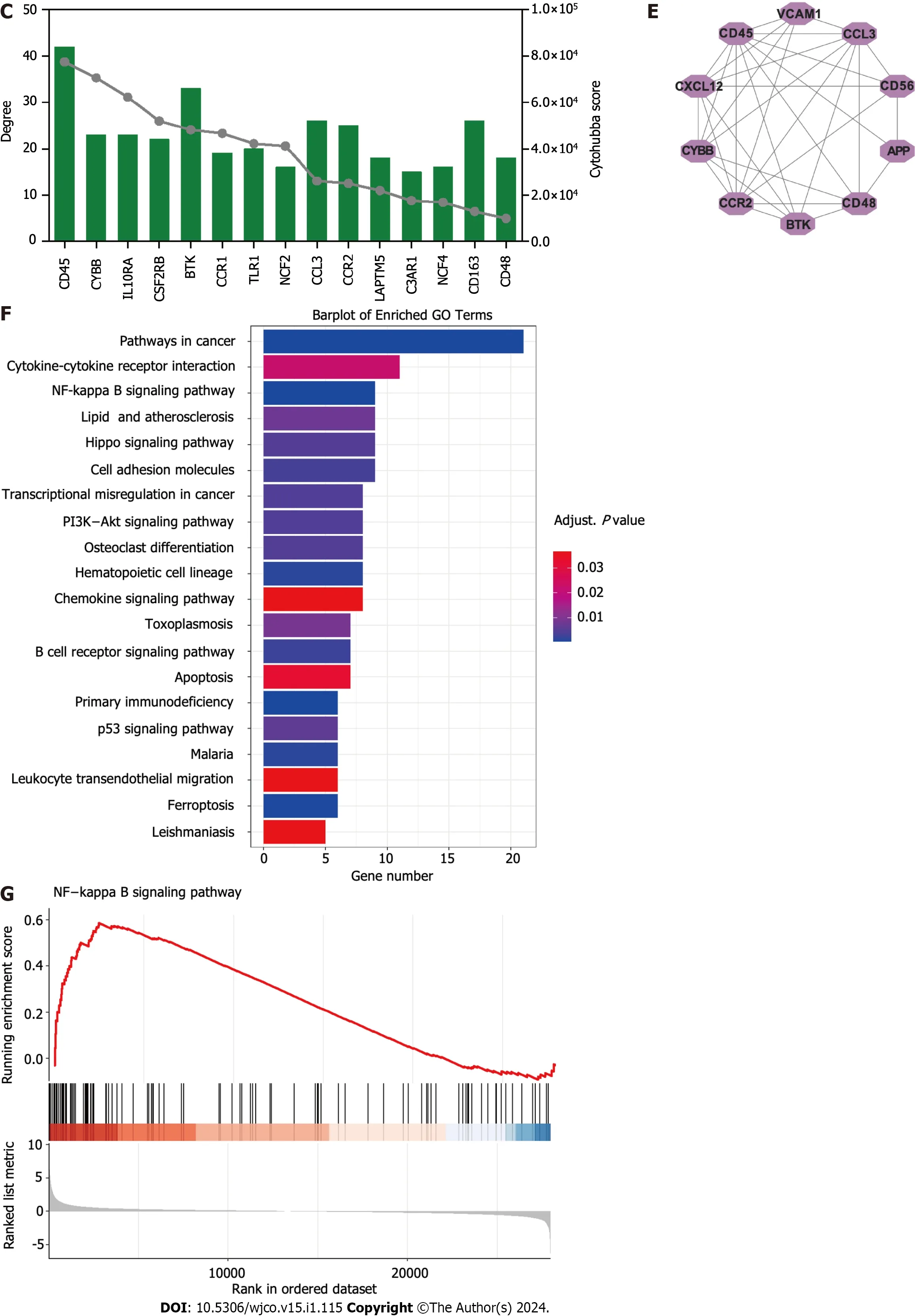
Figure 1 ldentification of hub targets and the Kyoto Encyclopedia of Genes and Genomes pathways.A: Venn diagram illustrating overlap of targets between GSE16558 and GSE116294 datasets;B: Target protein-protein interaction (PPI) network;C: Top 15 hub genes identified using CytoHubba plugin;D: Two primary clusters of PPI scored using themolecular complex detection plugin;E: Ranking of top 10 hub genes within PPI network;F: The Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis;G: Gene Set Enrichment Analysis of nuclear factor -κB signaling pathway.NK: Nuclear factor.
Potential targets involved in t(4;14) MM and PPI network:To better understand the therapeutic pathways involved in t(4;14) MM,we constructed a Venn diagram and identified 258 probable targets related to t(4;14) MM (Figure 1A).Subsequently,a PPI network containing these targets was constructed to elucidate the target interrelations.The PPI network comprised 175 nodes and 697 edges (Figure 1B).Three targets that lacked interactions with other targets were excluded from the assessment.The top five identified nodes were cd45,btk,ccl3,cdh1,and cxcl12,highlighting their pivotal roles in the network.
Identification of clusters and hub targets in the PPI network
Using the Cytoscape MCODE plugin,we identified two clusters with scores > 6 (Figure 1C).The first group,with 57 nodes and 343 edges,was centered on the seed gene,CD45,and was predominantly related to altered transcriptional regulation in cancer.The second smaller cluster,which included 15 nodes and 66 edges,hadTLR1as its seed gene and was primarily related to the negative regulation of apoptosis and signal transduction.Subsequently,the CytoHubba score was used to identify the top 15 genes (Figure 1D).Each of these 15 genes had a degree score >15,indicating comprehensive interactions among them.Combining insights from MCODE,MCC,and degree scores,we identified 10 central hub genes:cd45,vcam1,ccl3,cd56,app,cd48,btk,ccr2,cybb,andcxcl12(Figure 1E).
Functional enrichment analysis
To better understand the potential therapeutic targets of t(4;14) MM,we performed functional enrichment analysis of the 255 shared targets.Using the KEGG database,we identified 232 enriched signal pathway terms through enrichment analysis.With an adjustedPvalue of < 0.05,the top 20 pathways,including those related to cancer,cytokine-cytokine receptor interaction,NF-κB signaling,and the Hippo signaling pathway,were identified (Figure 1F).The GSEA for the NF-κB signaling pathway is illustrated in Figure 1G.Based on these findings,we focused on the NF-κB signaling pathway to elucidate the potential mechanisms related to t(4;14) MM.
Ivermectin identified as a potential drug using CMap
We used 258 DEGs as potential drug targets for t(4;14) MM and assessed the CMap database to identify small compounds that could serve as prospective drugs.Table 1 lists the top nine small-molecule drugs believed to hold therapeutic potential in countering the gene expression pattern of t(4;14) MM (with a cut-off score of < -80).Notably,ivermectin,which was listed among the identified small molecules,was considered for further investigation,as it is a high-ranking approved non-chemotherapeutic drug.Thus,we evaluated the potential inhibitory effects of ivermectin on t(4;14) MM cells.
Molecular docking
Molecular docking is a pivotal technique for the design of structure-based drugs.It facilitates the assessment of molecular interactions by determining the most favorable conformation between small-molecule targets and compounds.The 2D structure of ivermectin is shown in Figure 2A.In molecular docking simulations,ivermectin displayed strong binding affinity to all 10 identified targets,with docking energy scores below -7 kcal/mol (Figure 2B).Notably,CD45 and CYBB demonstrated the most potent binding,with binding energies of -10.2 kcal/mol and -9.9 kcal/mol,respectively.The 3D structural analysis revealedfavorable binding sites for ivermectin in both CD45 (Figure 2E) and CYBB (Figure 2F).Additionally,2D interaction diagrams indicated hydrogen bond formation between ivermectin and specific amino acid residues in CD45 (Glu1030 and Glu1003) and CYBB (Art73),along with hydrophobic interactions in CD45 (Figure 2C) and water-mediated interactions in CYBB (Figure 2D).These findings shed light on the binding mechanisms underlying the strong affinity between ivermectin and its targets,providing valuable insights for future drug design.
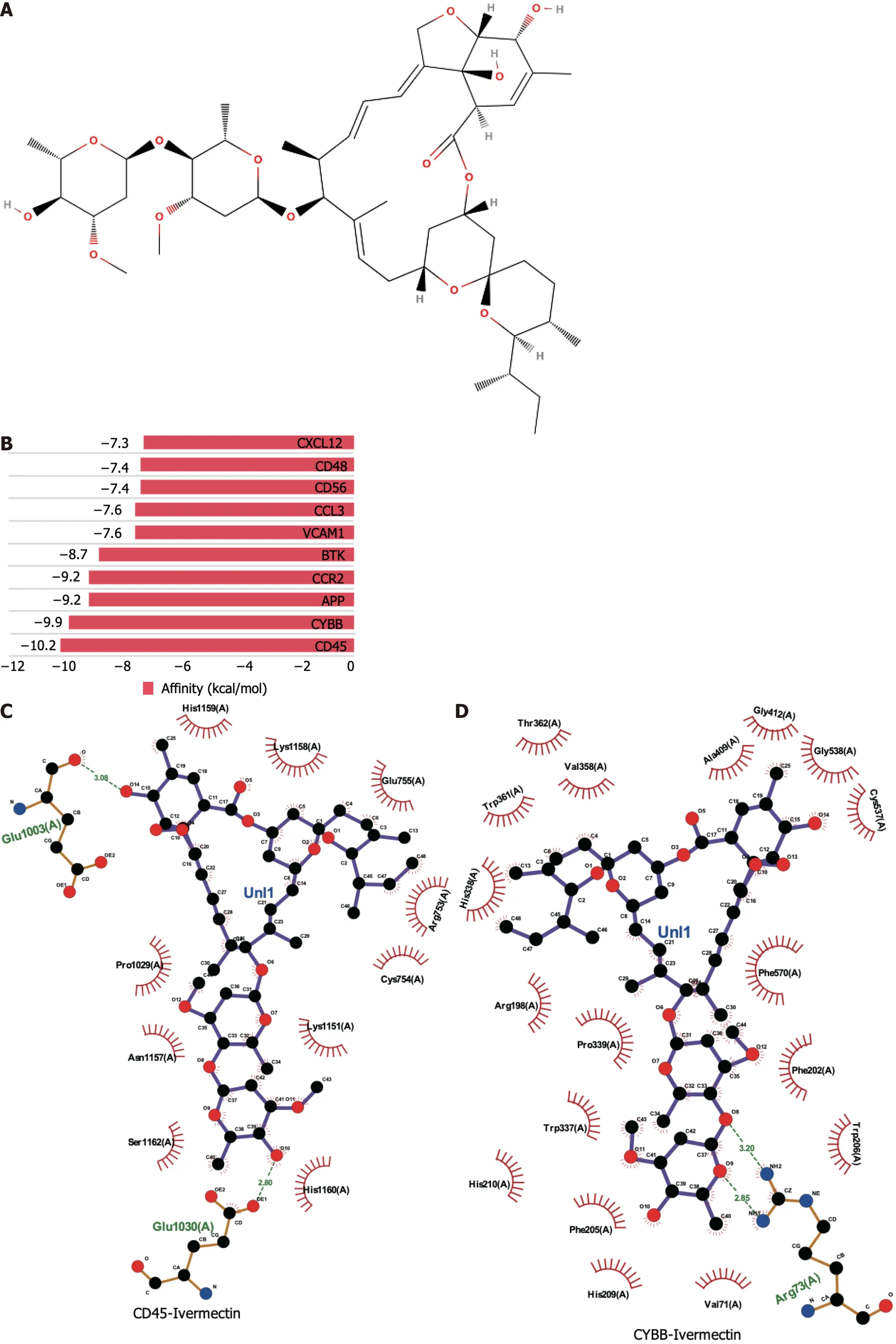
Figure 2 Molecular docking analysis of ivermectin with hub target genes. A: Chemical structure of ivermectin;B: Binding affinity of ivermectin with the top 10 hub-target genes.A longer bar indicates a lower binding affinity;C: 2D interaction diagrams of ivermectin with CYBB;D: 2D interaction diagrams of ivermectin with CD45;E: 3D docking structures and interactions of ivermectin with CD45;F: 3D docking structures and interactions of ivermectin with CYBB.
Ivermectin inhibits the proliferation of t(4;14) MM cells
We evaluated the impact of ivermectin on t(4;14) MM cell proliferation by subjecting cells to varying concentrations (0,4,6,8,10,and 20 µmol/L) of ivermectin and time intervals (24 and 48 h) of exposure.Cell viability,determined using CCK8,demonstrated a significant,concentration-and time-dependent decrease in cancer cell viability (Figure 3A and B).The half-maximum inhibitory concentration (IC50) of ivermectin was approximately 9.4 µmol/L in NCI-H929 cells.
Ivermectin induces apoptosis in t(4;14) MM cells
Drug-induced apoptosis is the primary mechanism underlying cancer cell death[18].To assess this,we analyzed the expression of the pro-and anti-apoptotic proteins BAX and BCL2,respectively,after treatment and further inspected the BAX/BCL2 ratio.We observed that ivermectin induced apoptosis in t(4;14) MM cells,as evidenced by the upregulation of pro-apoptotic protein BAX and a decrease in anti-apoptotic protein BCL2 levels (Figure 3C).The BAX/BCL2 ratio significantly increased (Figure 3D),and the intrinsic mitochondrial apoptotic pathway was activated,as indicated by elevated caspase-9,caspase-3,downstream effector caspase-3,and PARP expression levels (Figure 3E and F).Annexin VFITC/propidium iodide staining revealed a substantial increase in the proportion of apoptotic t(4;14) MM cells after ivermectin treatment compared with that of the control (Figure 3G and H),highlighting that the suppressive effect of ivermectin on t(4;14) MM cells was associated with apoptosis.
Ivermectin increases ROS accumulation and alters the mitochondrial membrane potential in t(4;14) MM cells
Mitochondria are widely known to be the intracellular source of ROS.These species can trigger oxidative damage,leading to a series of mitochondria-related events,including apoptosis[19].We detected a significant accumulation of ROS in t(4;14) MM cells treated with ivermectin compared with that in untreated controls (Figure 4A).We also observed a notable reduction in the mitochondrial membrane potential (Figure 4B and C),suggesting that ivermectin-induced apoptosis in t(4;14) MM cells was related to mitochondrial function.Collectively,these findings suggest a relationship between ivermectin-induced apoptosis in t(4;14) MM cells and altered mitochondrial dynamics.
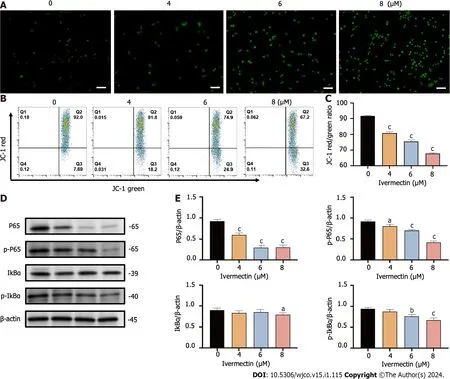
Figure 4 lvermectin induces apoptosis in t(4;14) multiple myeloma cells via mitochondrial and nuclear factor-κB signaling pathway. A: Visualization of intracellular reactive oxygen species (ROS) in t(4;14) multiple myeloma (MM) cells after treatment with ivermectin using 2′,7′-dichlorodihydrofluorescein (DCF) diacetate staining.ROS is represented by the green DCF fluorescence viewed under a fluorescence microscope at 200× magnification.Scale bar: 50 μm.B: Mitochondrial membrane potential in t(4;14) MM cells post-ivermectin treatment assessed using JC-1 staining;C: The data are shown as the means ± SD of three independent experiments;D: Expression patterns of p65,p-p65,p-IκBα,and IκBα in t(4;14) MM cells after exposure to ivermectin,analyzed by western blotting;E: The data are shown as the means ± SD of three independent experiments.aP < 0.05,bP < 0.01,and cP < 0.001 compared with the control group.
Ivermectin triggers apoptosis in t(4;14) MM cells via NF-κB signaling pathway
The NF-κB signaling pathway plays a crucial role in the onset and progression of the disease in t(4;14) MM,often exhibiting overactivation in these cells and promoting their survival[20].Our results revealed that ivermectin treatment substantially reduced both the protein expression and phosphorylation levels of NF-κB p65 in a dose-dependent manner (Figure 4D and E).Furthermore,the protein expression and phosphorylation levels of IκBα,a regulator upstream of NFκB,were also suppressed in t(4;14) MM cells post-ivermectin treatment.
DlSCUSSlON
MM is a multifaceted and incurable disease that displays vast heterogeneity in its clinical manifestations,genetic changes,therapeutic responses,and overall prognosis[2].Guidelines from bodies,such as the International Myeloma Working Group,recognize t(4;14) MM as a high-risk cytogenetic abnormality[21].The t(4;14) MM subtype continues to present a challenging prognosis despite advances in drug therapies[22].Therefore,there is an urgent need to develop new therapeutic drugs to treat t(4;14) MM.Recent studies have highlighted the potential of identifying molecular targets and therapeutic drugs for MM through expression profiling and functional enrichment analyses.Di Meoetal[23] identified ILT3 as an immunotherapeutic target for MM,whereas Mereuetal[24] revealed that UNC0642 increasedcarfilzomib sensitivity and counteracted drug resistance in MM cell lines.In this study,we identified the key genes and prospective therapeutic agents for t(4;14) MM using gene expression profile analysis.
The regulation of the immune response is fundamental for the development and progression of MM[25].We identified CD45,CD48,and CD56 as the key genes in t(4;14) MM.Compared with healthy bone marrow controls,patients with t(4;14) MM exhibited decreased CD45 expression levels,whereas CD48 and CD56 levels were notably increased.CD45,also known as protein tyrosine phosphatase receptor type C,was previously called the common leukocyte antigen.This protein is essential for modulating antigen receptor signaling,which is crucial for lymphocyte development,survival,and function[26].However,the role of CD45 in MM remains elusive.Evidence suggests thatCD45 expression decreases during MM progression[27].As mature MM cells are predominantly CD45-negative and have inactive SRC family kinases,they remain unaffected by elotuzumab[28].Moreover,MM cells in high-risk genetic categories tend to express reduced CD45 levels[29].CD48[30],a member of the signaling lymphocytic activation molecule family,plays a role in immune cell adhesion and activation.Although it is present in almost all MM plasma cells,it is absent from nonhematopoietic tissues.Van Ackeretal[31] identified CD48 as a promising molecular target for the therapy of MM antibody therapy.CD56,a neural cell adhesion molecule,is a glycoprotein present in neural and muscle tissues,as well as in myeloma cells.Cottinietal[32] observed its high expression levels in the t(4;14) MM cell line NCI-H929.CD56 boosts MM cell growth and influences adhesion to stromal cells.Collectively,these findings highlight the potential for a deeper evaluation of immune phenotypes for both diagnosis and treatment of t(4;14) MM.
In this study,we also utilized the CMap database to identify potential small-molecule drugs targeting t(4;14) MM.Our primary focus was on molecular docking and subsequent experimental validation of the predicted drug,ivermectin.Traditionally,ivermectin is a macrocyclic lactone antibiotic used to treat parasitic diseases[33].Recent studies have revealed its potential antitumor capabilities in various cancers,including breast[34] and pancreatic[35] cancer.Emerging evidence also suggests its synergistic lethal effects on MM cells when combined with proteasome inhibitors[36].However,a comprehensive understanding of the specific action mechanisms of ivermectin against MM,especially the t(4;14) subtype,remains elusive.Our findings demonstrate that ivermectin suppresses the growth of t(4;14) MM cells,as well as triggers apoptosis.
The NF-κB signaling pathway is crucial for driving cancer progression,angiogenesis,and shaping the tumor microenvironment[37].It orchestrates the production of pro-inflammatory cytokines,inflammatory mediators,and cell adhesion molecules,creating a favorable microenvironment for MM initiation and progression[38].Consequently,many leading anti-MM drugs indirectly target the NF-κB signaling pathway[39].Bortezomib hinders the proteasomal degradation of NF-κB and IκB proteins,inhibiting gene transcription activation[40].Our findings revealed a pronounced suppression of the NF-κB signaling pathway in ivermectin-treated t(4;14) MM cells (Figure 5).This suggests that ivermectin,by acting as an external signaling agent,inhibits the activation of this pathway,reducing anti-apoptotic effects and thereby enhancing cell apoptosis.
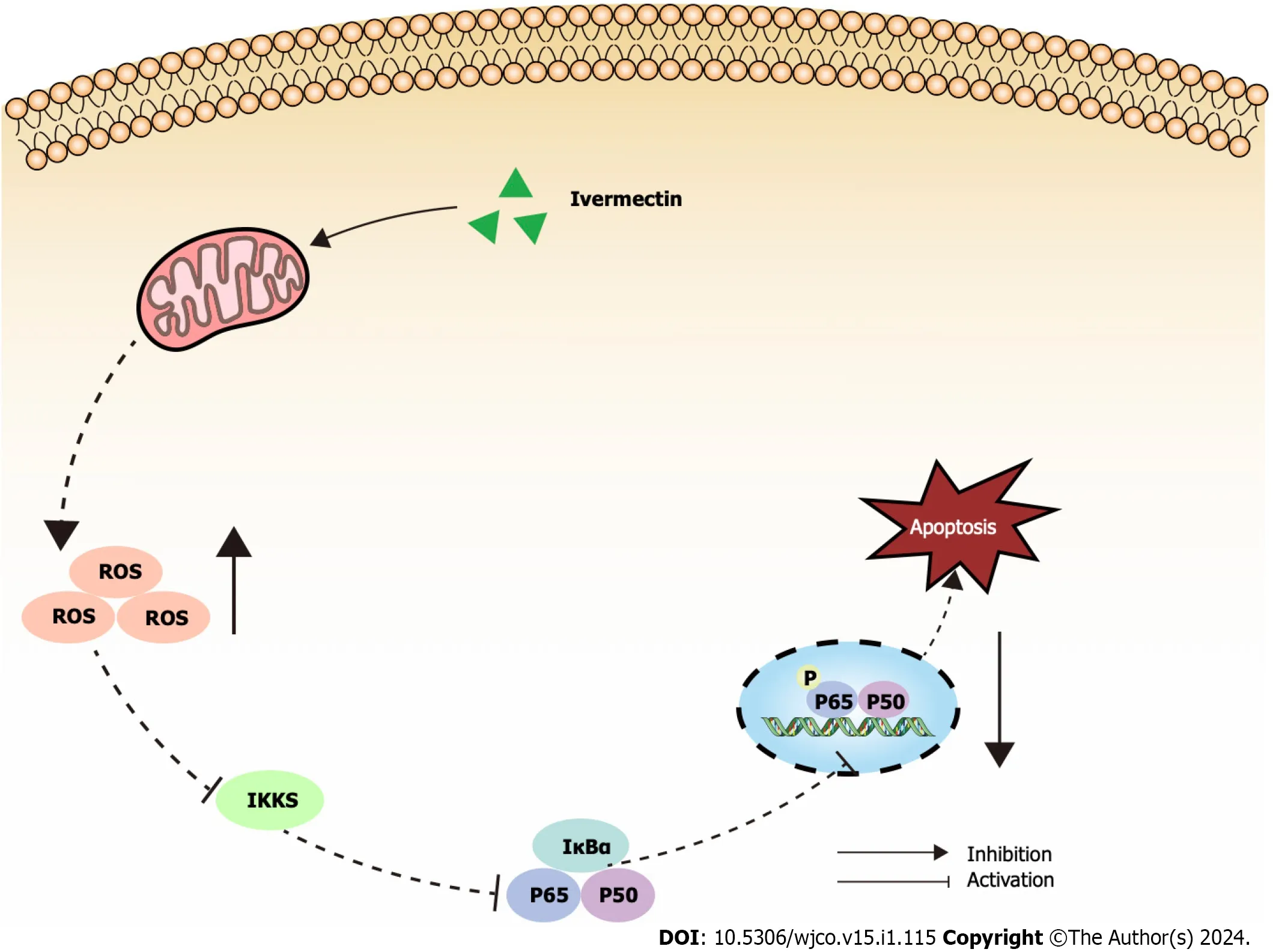
Figure 5 lllustrated overview of ivermectin promoting apoptosis in t(4;14) multiple myeloma cells. IκBα: Anti-inhibitor of NF-κB;IKKs: Inhibitors of kappa B kinase;ROS: Reactive oxygen species.
Mitochondria are essential for cell survival,functioning as the primary source of ROS and producing adenosine triphosphate (ATP) through oxidative phosphorylation[41].In a pathological state,mitochondrial dysfunction results inATP depletion[42].In the current study,treatment of t(4;14) MM with ivermectin led to elevated ROS levels and decreased cell membrane potential.These findings,along with previous reports,suggest that ivermectin exposure can induce oxidative stress and disrupt the mitochondrial balance,facilitating the apoptosis of t(4;14) MM cells.
There are also some limitations of the present study.Firstly,the sample size may be insufficient,potentially leading to selection bias.Secondly,regarding the experiments on ivermectin against t(4;14) MM,this study has only completed a portion of theinvitroexperiments,and further validation of its effectiveness and safety is still required ininvivoexperiments.Furthermore,additional genetic and experimental studies are needed to elucidate the mechanisms and functions of these key genes in the occurrence and development of t(4;14) translocated multiple myeloma.
CONCLUSlON
In conclusion,we utilized bioinformatics tools,molecular docking,and experimental validation to identify key genes and potential treatments for t(4;14) MM.Notably,we confirmed that ivermectin induced apoptosis in t(4;14) MM cellsviathe NF-κB signaling pathway.However,these insights require additional exploration and robust validation in further studies.
ARTlCLE HlGHLlGHTS
Research background
Multiple myeloma (MM) is a terminally differentiated B-cell tumor disease with a challenging prognosis.Specifically,the t(4;14) MM is categorized as a high-risk subtype within MM.
Research motivation
The t(4;14) MM tends to relapse,and currently,there is a lack of effective clinical treatment strategies.
Research objectives
This study aimed to elucidate the molecular basis of the t(4;14) MM and search for potential effective drugs through a comprehensive approach.
Research methods
The transcriptional characteristics of t(4;14) multiple myeloma were obtained from the Gene Expression Omnibus and subjected to gene ontology and pathway enrichment analysis.Utilizing the STRING database and Cytoscape,a proteinprotein interaction network was constructed,and core targets were identified.Connectivity Map identified potential small-molecule drugs,and these findings were validated through molecular docking analysis.One of these drugs,ivermectin,was further tested for its effects on t(4;14) multiple myeloma cells.
Research results
We identified 258 differentially expressed genes with enriched functions in cancer pathways,cytokine receptor interactions,the nuclear factor (NF)-kappa B signaling pathway.Ten key genes were pinpointed.Ivermectin emerged as a potential treatment.Invitro,ivermectin inhibited t(4;14) MM cell growthviathe NF-kappa B pathway and induced t(4;14) MM cell apoptosis.
Research conclusions
Ivermectin induced apoptosis in t(4;14) MM cellsviathe NF-κB signaling pathway.
Research perspectives
Our study offers valuable molecular insights for biomarker validation and potential drug development in t(4;14) MM diagnosis and treatment.
FOOTNOTES
Author contributions:Song Y and Lu XC conceived and designed the experiments.Zhang HJ,Geng J,and Song Y conducted the experiments and drafted the manuscript;Zhong LZ and Song X contributed to the techniques used and commented on the manuscript;Li HY performed the data analysis;Yang B and Lu XC assisted with revising the manuscript;All the authors reviewed the results and approved the final version of the manuscript.
Supported bythe National Key Research and Development Program of China,No.2021YFC2 701704;the National Clinical Medical Research Center for Geriatric Diseases,"Multicenter RCT" Research Project,No.NCRCG-PLAGH-20 230010;and the Military Logistics Independent Research Project,No.2022HQZZ06.
lnstitutional review board statement:This study does not involve research on humans/animals and does not include the initial version formally approved by the Institutional Review Board in the official language of the authors' country.
lnformed consent statement:This study does not involve clinical research and does not include the initial version of the informed consent form signed by all subjects and investigators.
Conflict-of-interest statement:The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data sharing statement:No additional data are available.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the original work is properly cited and the use is non-commercial.See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORClD number:Xue-Chun Lu 0000-0002-8937-2278.
S-Editor:Liu JH
L-Editor:A
P-Editor:Zhang XD
 World Journal of Clinical Oncology2024年1期
World Journal of Clinical Oncology2024年1期
- World Journal of Clinical Oncology的其它文章
- lnflammatory response in gastrointestinal cancers: Overview of six transmembrane epithelial antigens of the prostate in pathophysiology and clinical implications
- Uveal melanoma: Recent advances in immunotherapy
- Scinderin promotes glioma cell migration and invasion via remodeling actin cytoskeleton
- Prognostic and immunological roles of heat shock protein A4 in lung adenocarcinoma
- ldentification of the key genes and mechanisms associated with transcatheter arterial chemoembolisation refractoriness in hepatocellular carcinoma
- Predicting colorectal cancer prognosis based on long noncoding RNAs of disulfidptosis genes