
Biological factors driving colorectal cancer metastasis
2024-03-07 08:18ShuaiXingAnZhaoJinYuChenFuMinJieWeiLongHaiShen
Shuai-Xing An,Zhao-Jin Yu,Chen Fu,Min-Jie Wei,Long-Hai Shen
Abstract Approximately 20% of colorectal cancer (CRC) patients present with metastasis at diagnosis.Among Stage I-III CRC patients who undergo surgical resection,18% typically suffer from distal metastasis within the first three years following initial treatment.The median survival duration after the diagnosis of metastatic CRC (mCRC) is only 9 mo.mCRC is traditionally considered to be an advanced stage malignancy or is thought to be caused by incomplete resection of tumor tissue,allowing cancer cells to spread from primary to distant organs;however,increasing evidence suggests that the mCRC process can begin early in tumor development.CRC patients present with high heterogeneity and diverse cancer phenotypes that are classified on the basis of molecular and morphological alterations.Different genomic and nongenomic events can induce subclone diversity,which leads to cancer and metastasis.Throughout the course of mCRC,metastatic cascades are associated with invasive cancer cell migration through the circulatory system,extravasation,distal seeding,dormancy,and reactivation,with each step requiring specific molecular functions.However,cancer cells presenting neoantigens can be recognized and eliminated by the immune system.In this review,we explain the biological factors that drive CRC metastasis,namely,genomic instability,epigenetic instability,the metastatic cascade,the cancerimmunity cycle,and external lifestyle factors.Despite remarkable progress in CRC research,the role of molecular classification in therapeutic intervention remains unclear.This review shows the driving factors of mCRC which may help in identifying potential candidate biomarkers that can improve the diagnosis and early detection of mCRC cases.
Key Words: Colorectal cancer;Metastasis cascade;Cancer immunity;Genomic variation;Epigenetic instability;Lifestyle factor
lNTRODUCTlON
Colorectal cancer (CRC) is the second leading cause of cancer-related mortality and the third most common malignancy worldwide[1].Aging and an unhealthy diet and lifestyle are risk factors for CRC[2].In all metastatic CRC (mCRC) patients,the most common metastasis is found in the liver,followed by the lung and peritoneum,while metastases are found less frequently in the brain[3].The median survival duration after diagnosis of mCRC patients is only 9 mo.Although mCRC is traditionally considered to be caused by incomplete resection of tumor tissue or late-stage malignancy-related events,increasing evidence has demonstrated that the mCRC process can begin early during tumor development and that the genomic characteristics of these metastases typically impact cancer treatment.CRC patients exhibit high heterogeneity that manifests as different cancer phenotypes,which are classified in terms of morphological and molecular variations[4].Continuous accumulation of mutations leads to genotype diversity and promotes cancer evolution.Genomic variation,including driving point mutations,putative copy number alterations (CNVs),structural gene variants,and epigenetic instability,affects transcription factor expression,disrupts DNA methylation,and alters chromatin accessibility,which can lead to subclone divergence and thus supply energy for cancer evolution and metastasis (Figure 1).Through metastatic cascades,circulating cancer cells can move from an original tumor site to distant organs in the body and develop metastases[5].Therefore,identifying the driving biological factors of mCRC and measuring them as biomarkers or potential treatment targets for early intervention to prevent metastasis and even for late-stage treatment of advanced mCRC are extremely important.
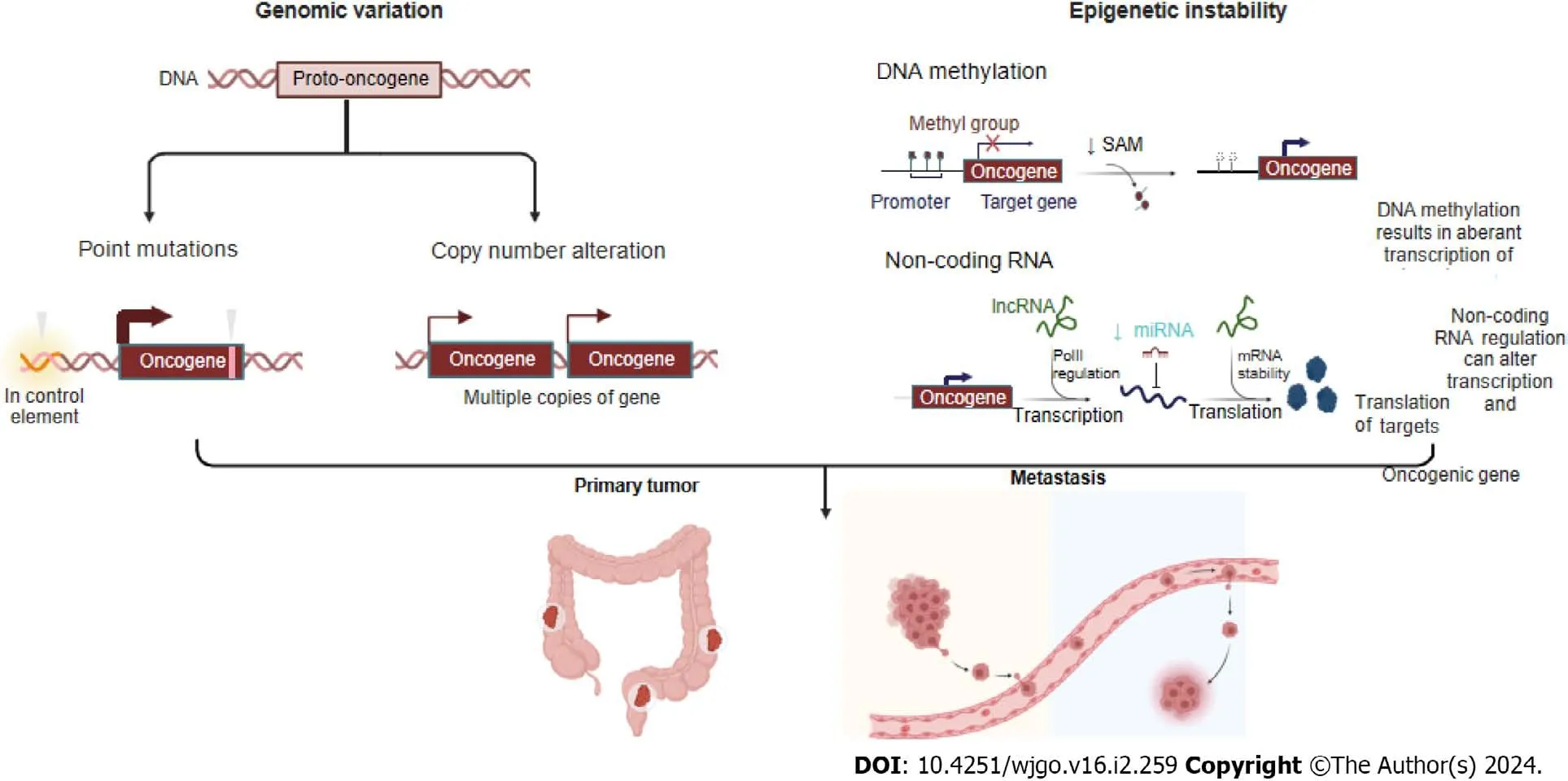
Figure 1 Different genomic events,including point mutations,copy number alterations,and epigenetic instability (such as disruption to DNA methylation and differential splicing based on noncoding RNA),can engender sub clonal diversity,thus providing fuel for primary tumor and its metastasis.Created with BioRender.com.
GENOMlC VARlATlON OF MCRC
Abundant molecular data on genomic variations have been obtained from both nonmCRC and mCRC samples,and some studies have focused on characterizing the molecular abnormalities in mCRC.
Driving mutations in mCRC
The mutational patterns and overall mutational burdens of primary and metastatic cancers are largely consistent,as shown by comparisons of CRC cohorts[6].Analysis of the mutation frequencies of certain genes in early stage tumors,mCRC tumors,and metastatic mCRC tumors from distal organs revealed that most of these mutations did not lead to significant differences,but a few were associated with tumor stage[4].Indeed,the accumulated alterations in these oncogenes play a crucial role in mCRC tumorigenesis and progression.However,whether mutations in these key oncogenes affect CRC metastasis remains unclear.
To identify oncogenes associated with the progression of metastases,Yaegeret al[7] compared the frequency of oncogenic variations in primary CRC tumors that did or did not develop metastasis in the TCGA and Memorial Sloan Kettering cohorts and identified 42 genes that were recurrently mutated to a significant degree in these cohorts.Alterations in TP53 were the only genomic mutations significantly enriched in mCRC samples,while alterations in FBXW7 were enriched in the early stage compared with mCRC samples,suggesting a potential protective role for FBXW7.A similar result was found by Vakianiet al[8] Among mCRC patients,TP53 mutations were notably more frequent,while BRAF mutations were less frequent.The frequencies of KRAS and PIK3CA mutations were not significantly different between primary CRC and mCRC samples.Liet al[9] conducted a study comparing a Chinese CRC (CCRC) patient dataset with a TCGA CRC dataset.Among CCRC patients,the proportion of mCRC patients was much greater than that in the TCGA CRC dataset (47.9%vs14.4%),and uniquely,SMAD4,a pivotal factor in the TGF-beta signaling pathway,tended to promote distant metastasis of CRC tumors[10] and was significantly more common in mCRC patients in the CCRC dataset than in mCRC patients in the TCGA dataset (20.0%vs6.6%;P=0.015)[9].A study by Huanget al[10] revealed no significant associations between mutations in APC or PIK3CA in mCRC patients.Subgroup analyses stratified by ethnicity showed that in Asian populations,KRAS,BRAF,and TP53 mutations,including lymph node and distant metastasis,were associated with mCRC,whereas only TP53 mutations promoted mCRC in Caucasians.Joet al[11] investigated the difference between mCRC primary tumors and mCRC metastatic tissues and identified a significant concordance of KRAS mutation status in 81.18% (9/11) of patients (P=0.03271).Only two patients showed intertumor heterogeneity.This intertumoral heterogeneity may be attributable to KRAS mutations early in disease progression from colorectal adenoma to malignant disease,leading to a continuous tumor growth advantage,whereas TP53 is mutated relatively late during CRC progression[12].
CNVs and mCRC
Increased CNVs,indicative of chromosomal instability (CIN),are strongly correlated with metastatic burden,and some chromosomes are significantly more unstable in some tumor metastases[13].However,in CRC,Nguyeet al[13] showed that CNV was not correlated with metastatic burden in colorectal adenocarcinoma.Similarly,another study showed that the most frequent CNV did not differ between mCRC and nonmCRC samples[9].However,several studies have reported exceptions;for example,a study by Casimiroet al[14] showed a greater frequency of MTDH amplification (copy number > 1.8) in mCRC patients with lung metastasis than in nonmCRC patients (17.4%vs100.0%,P< 0.001).Another prospective study showed that MYC was strongly amplified in mCRC patients with microsatellite stable (MSS) disease[13].A study by Liuet al[15] did not directly show CNV differences but classified chromosomal stable (CS) and CIN.The ratio of CIN to CS was found to be significantly increased in Stage III and Stage IV samples,which may explain why high-level CNV causes CIN and thus leads to CRC metastasis.
EPlGENETlC lNSTABlLlTY lN MCRC
Together,epigenetic instability and genetic alterations function to drive the progression of normal cells into cancer cells.Epigenetic instability is caused by several mechanisms,such as DNA methylation of cytosine bases in CG-rich sequences,which results in aberrant transcription of target genes;regulation of noncoding RNA,which can alter the transcription of oncogenic gene targets;and posttranslational modifications of histones,which regulate structural changes in packed DNA known as chromatin[16].
Methylation and mCRC
Many studies have attempted to identify genomic metastasis-promoting mutations.However,few metastasis-associated mutations have been found,and those that have been reported to date exhibit largely concordant mutation patterns and mutation burdens in both primary and metastatic cancers.Therefore,genomic mutations are not mediators of cascaderelated metastatic progression[6].However,several studies have shown unique features of a significantly greater frequency of CpG island hypermethylation in gastrointestinal (GI) adenocarcinomas than in non-GI adenocarcinomas[15],and marked differences in methylation were found in CRC patients.Therefore,Toyotaet al[16] named one CRC type,CpG island methylated subphenotype (CIMP),after observing its association with a unique molecular pathogenesis.It is widely accepted that aberrant gene hypermethylation is associated with tumor suppressor gene silencing,which leads to cancer formationviatranscriptional repression of these genes[17].Studying mCRC,Juet al[18] showed that the number of methylated genes was markedly greater in Stage I-III CRC than in the other two stage groups,namely,the stage IV primary mCRC and liver metastasis groups.However,when several studies compared primary CRC tumors and CRC liver metastases,the methylated phenotype was similar to that of primary cancer,which led researchers to conclude that most of the key DNA hypermethylation events associated with colorectal carcinogenesis likely occur before cancer cells spread to metastatic organs[19-21].
Furthermore,several studies have shown that differentially methylated genes,such as hypermethylated MGMT,which repairs alkylated DNA[19];hypermethylated TIMP3,which increases the EGFR signaling pathway by inhibiting MMPs and increasing the TNF signaling pathway by inhibiting ADAM[21];and hypomethylated nuclear element-1 (LINE-1) enables the inadvertent activation of methylation-silenced proto-oncogenes in mCRC[20,22].In summary,aberrantly methylated genes play important roles in the initiation and progression of mCRC and can even interfere with drug responses[15].
Noncoding RNAs and mCRC
Previously,98% of noncoding sequences were considered useless,but increasing evidence has shown that these RNA transcripts strongly impact different physiological and pathological processes[23].To date,more than 1400 miRNAs have been identified,accounting for 2%-5% of the entire human genome and regulating 30% of human gene expression[24].Recently,several studies have been performed to determine the regulatory roles of noncoding RNAs (ncRNAs) in different steps of the colorectal metastasis cascade and how ncRNAs coordinate a series of pathological events.
A study by Mokutaniet al[25] showed that downregulation of miR-132 by targeting downstream ANO1,which is a key oncogenic factor,contributed to the CRC metastatic cascade.A study by Huet al[26] showed that low expression of miR-744,which is a target of Notch1,was positively correlated with TNM stage;in contrast,overexpression of miR-744 significantly inhibited the proliferation and invasion of CRC cells.A study by Yanet al[27] indicated that miR-520d-5p was significantly downregulated by targeting CTHRC1,which is involved in the progression and metastasis of CRC.Additionally,it is regulated by SP1 and affects the epithelial-mesenchymal transition (EMT) by inactivating the phosphorylation of Erk1/2.A study indicated that significantly upregulated miR-425 and miR-576,which target PTEN,were key factors in CRC liver metastasis[26,28].
Lianget al[29] reported that the long non-coding RNA (lncRNA) RPPH1 was markedly upregulated in CRC tissues and RPPH1 overexpression induced EMT in CRC cells by interacting with TUB3 to prevent its ubiquitination,an outcome that has been associated with advanced TNM stage and poor prognosis.Two studies identified functional roles for CYTOR (a lncRNA also known as LINC00152) in CRC progression.The first of these two studies,conducted by Wanget al[30],showed that CYTOR forms a heterotrimeric complex with the RNA-binding proteins NCL and Sam68 through EXON1,which activates the NF-κB signaling pathway,thus promoting EMT and CRC metastasis.The second study,conducted by Lvet al[31],showed that CYTOR mediates the binding of ENO2 to large tumor suppressor 1 (LATS1) and competitively inhibits the phosphorylation of Yes-associated protein 1 (YAP1),which ultimately triggers the EMT and CRC metastasis.
MSI and mCRC
A series of studies led to the understanding that deficient DNA mismatch repair (dMMR) causes microsatellite instability (MSI);importantly,most sporadic MSI-type CRCs are derived from CIMP-type CRC[15].A study by Tienget al[32] showed that only 4% of mCRC patients exhibited the MSI genotype,a significantly lower frequency than that reported for nonmCRC patients,which can be explained by the MSI subtype showing a much lower tendency to metastasize.More detailed data from the study by Liuet al[15] showed that in patients with Stage IV CRC,the percentage of patients with MSI was 28%,and that of patients with MSS was 72%,with the same ratio of MSI-to-MSS in patients with Stage III CRC.These ratios were significantly different from those identified in patients with Stage I or II CRC;42% presented with the MSI type,and 58% presented with the MSS type.Accordingly,MSI-type patients,including mCRC and nonmCRC patients,exhibited a longer OS[15].Moreover,in the lower GI tract,CIMP-H-and MSI-type tumors were largely absent in the descending colon compared with their incidence in the upper GI tract[33].
Recent studies showing the efficacy of immune checkpoint inhibitor therapy in dMMR-MSI-high CRC patients have indicated promising effects,but its use in patients with the MSS subtype has either been unsuccessful or not widely explored[34].The likely explanation for the discriminative efficacy of this approach in patients with the MSI genotype is because it is associated with the next-highest IFN-γ expression signature[35],and diverse immune signatures,such as high expression levels of the checkpoint protein CD276[15],can lead to the accumulation of mutations and subsequent immunogenic neoantigens[36],inducing high cytotoxic T cells (CTLs) engagement and better clinical outcomes[37].Mechanistically,the group of patients with MSI-type CRC harbored lower WNT expression signatures than did the other CRC groups,a finding that may have been attributable to reduced metastasis[15].Our understanding is that among mCRC patients,a much greater incidence of the MSS subtype was identified,and patients with this subtype exhibited a lower immune response (Figure 2).
MCRC AND METASTATlC CASCADES
To metastasize,a series of metastatic cascades,such as invasive migrationviathe circulatory system,extravasation,distal cell seeding,dormancy and reactivation,characterize cancer cells,with each step triggered by specific functions.In these cascades,EMT,angiogenesis,hypoxia,and circulating tumor cells (CTC) and exosomes are the key factors (Figure 2).
mCRC and EMT
The EMT is a critical process during cancer metastasis in which epithelial cells acquire the phenotype of mesenchymal stem cells.Cells undergoing the EMT are at the invasive front of a cancerous tumor,and the EMT drives cancer cell migration,invasion,and metastatic spread[38].Multiple signaling pathways,such as the TGF-β,BMP,RTK,Wnt/βcatenin,Notch,Hedgehog,and STAT3 pathways as well as ECM-mediated and hypoxia-regulated pathways,are involved in the EMT[39].Members of the Snail family of transcriptional regulators,namely,Snail1 and Snail2,and the zinc finger transcription factors Zeb1 and Zeb2 also make critical regulatory contributions to the EMT[40].
Several studies on mCRC have shown the impact of genetic and relevant signaling pathways on EMT and the role of EMT in cell migration from the original site to metastatic sites (Table 1)[41-49].

Table 1 Epithelial-mesenchymal transition signaling pathways
mCRC and angiogenesis
Angiogenesis is a hallmark of cancer and is closely related to tumor growth,cancer cell metastasis and invasion,prognosis,and recurrence.Angiogenesis,which involves the formation of new vessels from preexisting vessels,is critical for the progression of both primary and metastatic cancer[50].At metastatic sites,angiogenesis enables malignant cells to repeat the entire sequence of events required for further metastasis[51].
Vascular endothelial growth factor A is a major proangiogenic factor that is associated with metastasis formation and poor prognosis in CRC patients[52].Many studies have shown that the VEGF signaling pathway has high therapeutic value because it regulates angiogenesis in CRC patients.Noguéset al[53] reported that VEGF serum levels in mCRC patients before surgery were significantly greater than those in nonmCRC patients.Therefore,antiangiogenic therapeutic strategies are important and effective tools for improving outcomes in patients diagnosed with mCRC in specific settings[54].Many drugs targeting VEGF,platelet-derived growth factor,fibroblast growth factor,and their receptors are marketed or are under development[55].
mCRC and hypoxia
The metastatic process exerts strong selective pressure on cancer cells,and metastatic cancer cells develop high oxidative stress.However,the Warburg effect helps cancer cells minimize oxidative stress by inhibiting mitochondrial oxidative metabolism,thereby promoting metastatic spread[56].As a hallmark of cancer,the Warburg effect facilitates CRC metastasis by promoting angiogenesis,promoting cancer-associated fibroblast formation,and suppressing the immune system,and it can also lead to drug resistance[57,58].
Qiet al[59] analyzed a total of 1730 CRC samples and were able to classify most of them into two subgroups: a hypoxia subgroup and a normoxia subgroup.They found that hypoxia was associated with poor prognosis in CRC and was closely associated with activation of the RAS signaling pathway independent of KRAS mutation.Furthermore,hypoxia promoted M2 macrophage infiltration and was associated with poor outcomes[59].In addition,other researchers have shown that hypoxic cell-derived miR-135a-5p exosomes promote CRC liver metastasis by suppressing the kinase 2-YAP1-matrix metalloproteinase 7 axis[60],and hypoxic cell-derived circ-133 exosomes promote cancer metastasis by acting on the miR-133a/GEF-H1/RhoA axis[61].
CTCs and exosomes
Clearly,only a small portion of CTCs can undergo metastasis,while multiple mechanisms in heterogeneous CTCs facilitate their metastatic potential by driving CTC interactions with immune and stromal cells[62].First,Chiuet al[63] showed that,compared with carcinoembryonic antigen (CEA) alone,CTC detection increases the power of the area under the receiver operating characteristic curve for predicting mCRC (0.7800vs0.8378).Surprisingly,Leet al[64] showed that CTCs were not associated with the size or number of metastases,as determined after previously administered drug therapy,or disease-free survival (DFS).Only tumor marker-positive thoracic lymph nodes were associated with the presence of CTCs in pulmonary venous blood,and CTCs were present in all patients (Fisher’s exact test,P=0.02).Moreover,researchers have shown that the results of overall and DFS analyses are not different regardless of whether CTC marker expression or CTC number is considered[64].However,some studies have shown that heterogeneous CTCs promote metastasis.Gkountelaet al[65] demonstrated that CTC clustering altered DNA methylation,specifically hypomethylation of the binding sites of OCT4,NANOG,SOX2,and SIN3A,which is similar to what has been observed in embryonic stem cells,and promoted stemness and metastasis.Hamidet al[66] showed that the potential target gene AKT1 was expressed at a significantly (P=0.0129) greater level in single CTCs from Stage III or IV samples than in early stage samples.
Exosomes,as primary communication mediators,are extracellular vesicles with multiple biological functions[67].Exosomes play important roles in the development of mCRC,including enhancing tumorigenicity;promoting angiogenesis,cancer cell proliferation,and endothelial cell migration;and establishing an immunosuppressive environment[68].Several studies have demonstrated that tumor-derived exosomes and their functions in CRC metastasis.For example,miR-27b-3p-enriched exosomes increase the permeability of blood vessels and facilitate CTC generation[69],and miR-25-3p-,miR-130b-3p-,miR-425-5p-,miR-934-,and RPPH1-enriched exosomes induce M2 polarization of macrophages and promote cancer metastasis[29,70,71].In addition,miR-200b-enriched exosomes are transferred to a targeted cell to increase CRC cell proliferation by directly targeting the 3′-UTRs of p27 and RND3[72],and ANGPTLIenriched exosomes reprogram Kupffer cells and decrease MMP9 expression to hinder vascular leakiness in liver premetastatic niches[73].HSPC11-enriched exosomes reprogram lipid metabolism in cancer-associated fibroblasts (CAFs) to facilitate premetastatic niche formation and liver metastasis[74].circLONP2 exosomes modulate miR-17-5p intracellular maturation and intercellular transfer and are subsequently internalized by adjacent cells to increase their metastatic ability[75],and circPACRGL exosomes play an oncogenic role in CRC proliferation and metastasis[76].
In addition to tumor cell-derived exosomes,nontumor cell-derived exosomes also play roles in CRC metastasis.Huet al[77] showed that CAF-derived miR-92a-3p exosomes promoted cell stemness,the EMT,metastasis and chemoresistance in CRC cells by activating the Wnt/beta-catenin signaling pathway and inhibiting mitochondrial apoptosis through FBXW7 and MOAP1 inhibition.Renet al[78] also showed the same functions in CAF-derived H19 exosomes by activating the Wnt/beta-catenin signaling pathway and acting as competing endogenous RNA sponges for miR-141,while miR-141 inhibited the stemness of CRC cells.In addition,several researchers have initiated exosome therapy studies with CRC samples.MiR-155-enriched exosomes,also called dendritic cell (DC) immunotherapy,induce antitumor immune responses and prolong survival in a CRC mouse model by increasing the expression of principal cytokines[79].
MCRC AND lMMUNlTY
The cancer-immunity cycle includes neoantigen generation during tumorigenesis,DC processing and antigen presentation,which activate effector T cells that respond to cancer antigens and kill targeted cancer cells (Figure 3).However,in cancer patients,the cancer-immunity cycle is not optimal due to low neoantigen levels and subsequent failure to activate T cells or suppress immunity[80].It is widely accepted that immune cells in the tumor microenvironment (TME) significantly affect the progression of CRC at the primary tumor site and metastatic sites[81].For CRC patients,the immunoscore,which is based on quantified immune cell infiltration,has been shown to be superior to the current staging system[82,83],and immunoscore diagnostic kits have been approved for CRC patients[84] (Figure 3).
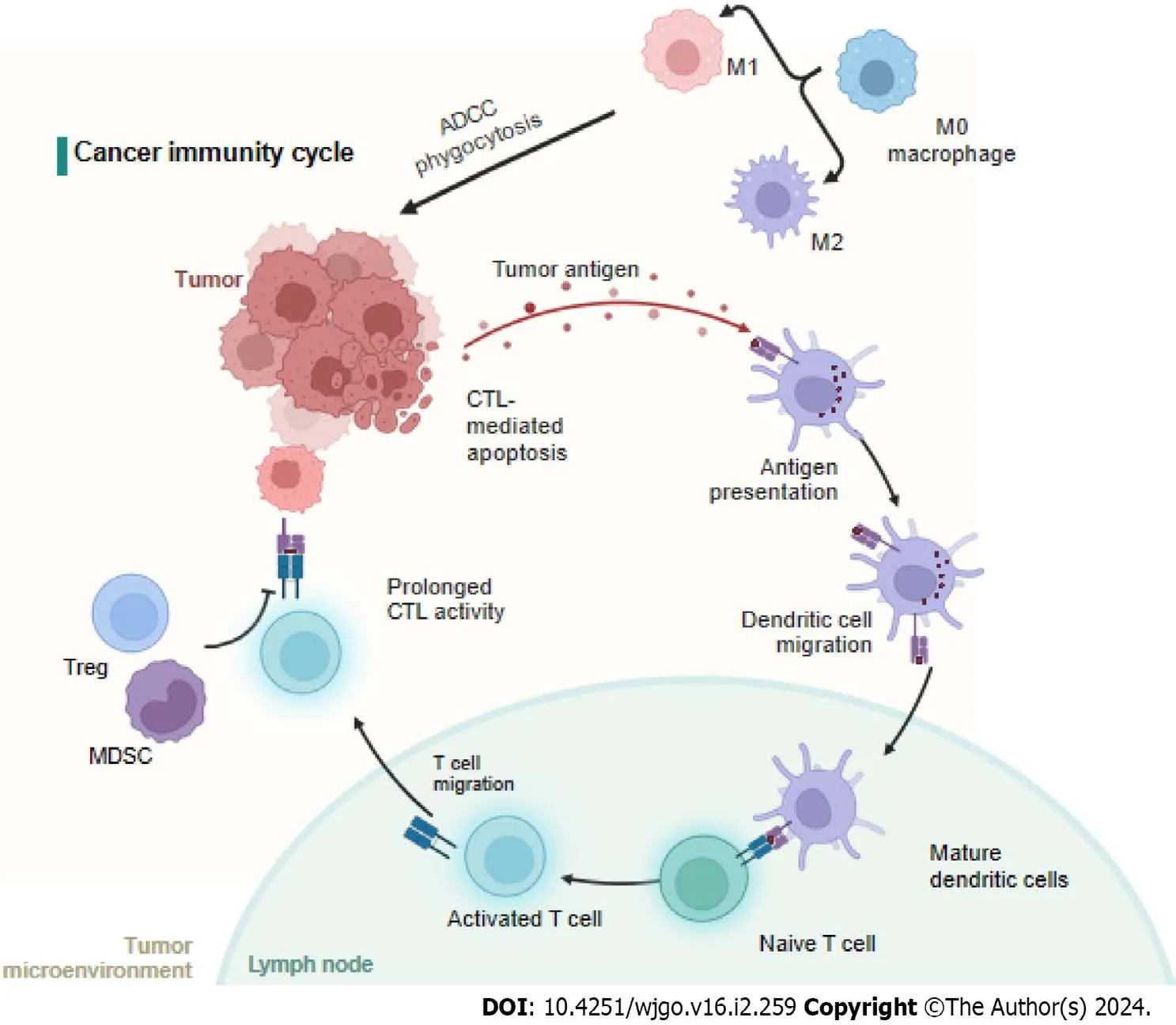
Figure 3 Neoantigens created by oncogenesis are released and captured by dendritic cell for processing. Dendritic cells present the captured antigens on MHCI and MHCII molecules to T cells,resulting in the priming and activation of effector T-cell responses against the cancer-specific antigens.Finally,the activated effector T cells move toward and infiltrate to the tumor bed.MDSC: Myeloid-derived suppressor cells;ADCC: Antibody dependent cell mediated cytotoxicity;CTC: Circulating tumor cells;CTL: Cytotoxic T cell.Created with BioRender.com.
mCRC and innate immunity
M0 macrophages are polarized to the M1 subtype through the action of LPS and IFN-γ or to the M2 subtype after induction by IL-4,IL-10,and IL-13[85-87].Studies have shown that the M1 subtype can increase immunity by recruiting CTLs and inducing cancer cell apoptosis through phagocytosis,antibody dependent cell mediated cytotoxicity,and the release of TNF and nitric oxide.In contrast,the M2 subtype leads to angiogenesis,the EMT,and immunosuppression to promote CRC metastasis[88].Interestingly,the number of circulating or tumor-infiltrating NK cells is inversely associated with metastasis in CRC patients[89].However,another study showed that NK cells rarely infiltrated tumors,but a high number of tumor-associated NK cells correlated with good clinical outcomes in CRC patients[90].Moreover,the number of tumor-infiltrating NKT cells was positively correlated with good clinical outcomes in CRC patients[91].
mCRC and adaptive immunity
By presenting tumor antigens,DCs induce specific polarization of T lymphocytes into different subsets of cells.However,the mechanisms by which these DCs enhance invasion are unclear.However,as observed in CRC,tumor-infiltrating DCs exhibit distinct patterns of tumor infiltration according to their maturation status,which can partially explain their highly variable prognostic value[92].One study showed DC-related outcomes in which an increased density of CD208-positive DCs was associated with worsened disease outcomes in CRC patients[83].
CTLs are important components of antitumor immunity,and the number of CTLs among infiltrating cells correlates with low recurrence in CRC patients[93,94].Similarly,Lazaruset al[95] showed that an increased number of CTLs is more frequently associated with epithelial cells in the tumor microenvironment of MMR-deficient mCRC and prolongs overall survival.Bindeaet al[96] reported that increased T-cell infiltration was associated with reduced metastasis,which likely reflects ongoing adaptive immune pressure on tumor development and spread.In general,CD4+helper T cells modulate the positive effects of the cancer immune response by secreting cytokines[97].T-helper 1 (TH1) cells enhance CTL effectors,and T-helper 17 (TH17) cells exhibit immunosuppressive effects,leading to poor clinical outcomes[98].Amicarellaet al[99] demonstrated that IL-17 derived from TH17 cells promoted the production of protumor genetic factors,while IL-8 derived from TH17 cells induced cytotoxic CCR5+CCR6+CD8+T-cell infiltration into the CRC tumor microenvironment and recruited neutrophils,CC-chemokine ligand 5 (CCL5),and CCL20.Moreover,two studies have shown that,in mCRC samples,the T-cell density is lower and the B-cell density is greater than those in nonmCRC tumor microenvironments[100,101].
Regulatory T cells (Tregs) characterized by CD25 and FoxP3 expression are considered potent mediators of immunosuppression.However,in CRC,Tregs have complex effects on the TME,and evidence suggests that their protumorigenic and antitumorigenic functions are context specific[81].The number of myeloid-derived suppressor cells in peripheral blood was positively associated with cancer stage in CRC patients[102,103].Furthermore,fully differentiated neutrophils with increased granule density and increased CD66b+neutrophils were associated with better outcomes in CRC patients[104,105].
MCRC AND LlFESTYLE FACTORS
In addition to internal biological factors,external lifestyle factors,such as overweight/obesity,physical inactivity,cigarette smoking,alcohol consumption and inappropriate dietary patterns,also influence metabolism,cell survival,tumor progression and metastasis in CRC patients[106].
mCRC and body mass index
A high body mass index (BMI) is a convincing risk factor for the development of CRC,and the overall CRC risk is estimated to increase by 3% for every five kilograms of weight gain[107].Among the mCRC patient cohort,63% and 27% were overweight with a BMI > 25 kg/m2and BMI > 30 kg/m2,respectively[108].Mechanistically,this could be explained by the fact that the adipose tissue of patients releases more unfavorable factors,such as TNF-α,IL-1,IL-6,IL-7,and IL-8,which inhibit apoptosis,promote oxidative stress,suppress the immune response,and reduce the activity of the IGF-1 axis;these factors are also associated with cancer development and progression[109].Another study showed that overweight and obese mCRC patients who were receiving therapies targeting VEGF had poorer outcomes with bevacizumab[108].However,a low BMI is associated with an increased risk of progression and death among the patients enrolled in mCRC trials,while there is no increased risk for an elevated BMI,in contrast to the adjuvant setting in mCRC patients[110].
mCRC and dietary intake
Studies have shown that regular consumption of red and processed meat is an important risk factor that may increase the risk of mCRC by approximately 17% for every 100 g portion of red meat and approximately 18% for every 50 g of processed meat eaten daily;moreover,it was shown that high consumption of dietary fiber could reduce the risk of CRC development by up to 50%[109].However,more than 80% of Stage III and metastatic CRC patients fail to meet the US FDA recommended daily intake of vegetables,fruits,and milk products[107].Mechanistically,harmful substances,such as heterocyclic amines and polycyclic aromatic hydrocarbons,generated from grilled and roasted meat have the potential to cause point mutations (deletions,insertions,and substitutions).Similarly,nitrosamines and nitrosamides are potent carcinogenic agents that can react with DNA.In contrast,the potential mechanism of the protective effect of fiber consumption on CRC development occurs through reducing contact between carcinogenic substances and the colonic epithelium as well as stimulating the growth of beneficial gut microbiota.
mCRC and lifestyle habits
Tobacco smoke is an established risk factor for the development of many types of cancer,including CRC.Smoking cigarettes increases the risk of developing CRC by 2-to 3-fold compared with nonsmokers[111].The mCRC cohort represented 9% of the smokers,while the Stage III cohort represented 10% of the smokers;these two cohorts were not significantly different[107].Tobacco contains a mixture of thousands of chemicals,more than 60 of which are wellestablished carcinogens that are known to damage DNA and lead to mutations.
Alcohol intake is another contributor to CRC development.However,the data show that CRC risk has no significant correlation with light to moderate alcohol consumption[112].Although approximately half of the patients in the two cohorts (47% of Stage III patients and 43% of metastatic patients) reported no alcohol intake during the 3 mo prior to questionnaire completion[108],the risk of heavy drinking is remarkable,and people who drink more than 4 times have a 52% risk of developing CRC[113].Furthermore,a Canadian study reported that among subjects who consumed an alcohol beverage at least once a week for 6 mo or longer,those with a BMI > 30 had an overall CRC OR of 2.2[114].Mechanically,alcohol oxidation and nonoxidative metabolism and the formation of byproducts,such as ROS and metabolites,can lead to a constellation of genetic,epigenetic,cell signaling,and immune processes[115].Similarly,decreased miR-135 expression in response to ethanol exposure plays a role in colon carcinogenesis and enhances metastasisviaAPC suppression[116,117].It has also been shown in an HCT116 cell model that ethanol inactivates GSK3β,leading to increased nuclear translocation of β-catenin and induction of cancer stem cell metastasisviathe production of MCP-1/CCR-2[118].
CONCLUSlON
Despite remarkable progress in CRC research,the role of molecular classification in therapeutic intervention has not been fully elucidated.This review highlights the driving factors of mCRC and may help in identifying potential candidate biomarkers that can improve the diagnosis and early detection of mCRC,thereby prolonging the overall survival and clinical outcomes of CRC patients.Using molecular alterations to predict CRC risk is a promising approach,but further research is needed to determine whether aberrant mutations,methylation patterns,CNVs,epigenetic marks,immune cell infiltration,and lifestyle factors can be used as reliable and accurate indicators of mCRC risk.
FOOTNOTES
Author contributions:An SX and Yu ZJ designed the article;An SX and Fu C performed the literature search and drafted the work;Shen LH and Wei MJ revised the work;and all authors are responsible for obtaining written permission to use any copyrighted text and/or illustrations.
Conflict-of-interest statement:The authors declare that they have no conflicts of interest.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the original work is properly cited and the use is non-commercial.See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORClD number:Shuai-Xing An 0009-0003-4657-590X;Min-Jie Wei 0000-0002-0404-7098;Long-Hai Shen 0000-0001-9665-2289.
S-Editor:Chen YL
L-Editor:A
P-Editor:Zhao YQ
 World Journal of Gastrointestinal Oncology2024年2期
World Journal of Gastrointestinal Oncology2024年2期
- World Journal of Gastrointestinal Oncology的其它文章
- Targeting oxidative stress with natural products: A novel strategy for esophageal cancer therapy
- Multifaceted role of microRNAs in gastric cancer stem cells: Mechanisms and potential biomarkers
- Effect of different anesthetic modalities with multimodal analgesia on postoperative pain level in colorectal tumor patients
- Prognostic value of circulating tumor cells combined with neutrophil-lymphocyte ratio in patients with hepatocellular carcinoma
- Systemic lnflammation Response lndex and weight loss as prognostic factors in metastatic pancreatic cancer: A concept study from the PANTHElA-SEOM trial
- Prohibitin 1 inhibits cell proliferation and induces apoptosis via the p53-mediated mitochondrial pathway in vitro