
盐酸贝那普利片及活性代谢物在中国健康受试者中的生物等效性研究
2024-03-04 00:37段舟萍彭洪薇魏筱华崔建鑫
南昌大学学报(医学版) 2024年1期
段舟萍,彭洪薇,李 蒲,魏筱华,崔建鑫,韩 盈
(1.南昌大学第一附属医院a.临床研究中心; b.药学部,南昌 330006; 2.科文斯医药研发有限公司,上海 200001)
盐酸贝那普利片是临床常用的血管紧张素转化酶抑制剂(angiotensin converting enzyme inhibitors,ACEI)类抗高血压药,该药口服后在体内迅速被吸收,代谢为活性物质贝那普利拉,通过减少醛固酮分泌,降低血管阻力,增高血浆肾素活性,还可抑制缓激肽的降解,从而发挥降压作用[1-2]。本研究旨在考察国产10 mg盐酸贝那普利片制剂在健康人群的药代动力学参数,并评价其与原研药品的参比制剂的生物等效性。
1 材料与方法
1.1 药品与试剂
受试制剂(test,T):盐酸贝那普利片,规格:10 mg·片-1,批号:DGM20160601,由深圳信立泰药业股份有限公司生产;参比制剂(reference,R):盐酸贝那普利片(商品名:Cibacen®),规格:10 mg·片-1,批号:3785401,由原研MEDA Pharma GmbH &Co.KG生产。贝那普利盐酸盐的标准品批号为2-FJ-123-1,纯度为98.0%;贝那普利拉标准品批号为1-JQW-132-1、纯度为98.0%,内标物贝那普利-d5盐酸盐标准品批号为1368-087A1、纯度为99.4%,贝那普利拉-d5标准品批号为7-JQW-116-1、纯度为98.5%,均由TLC Pharmaceutical Standards生产。实验用水为超纯水,乙腈、甲酸、甲醇、二甲基甲酰胺和乙酸乙酯等化学试剂均为HPLC级或化学纯级。
1.2 仪器
Nexera UHPLC LC-30A高效液相色谱仪,由日本岛津公司生产;Sciex API 5500三重四级杆质谱仪,由美国Series公司生产;微量电子天平MX5,由美国梅特勒-托利多公司生产。
1.3 受试者选择
本研究经南昌大学第一附属医院伦理专业委员会批准(伦理号:2017临伦审087号;餐后及空腹试验的药物临床试验登记号分别为CTR20181024、CTR20180633),所有志愿者在参加试验前均签署知情同意书。本研究空腹组共入组30例健康志愿者,均为汉族,男10人,女20人,年龄(23.90±4.14)岁,身高(161.02±8.33)cm,体重(57.13±8.59)kg,BMI(21.92±1.77)kg·m-2;餐后组共入组30例,28人为汉族,2人为满族,男14人,女16人,年龄(23.4±4.13)岁,身高(162.00±8.14)cm,体重(57.12±7.18)kg,BMI(21.70±1.26)kg·m-2。
入选及排除标准参考文献[3]。入选标准:1)年龄≥18周岁;2)男性体重不低于50 kg,女性体重不低于45 kg,BMI在19~26 kg·m-2;3)经病史询问、全身体检以及实验室检查证明为健康者。排除标准:1)已知对贝那普利或其他ACEI制剂有过敏史者;2)体格检查异常且有临床意义者;3)生命体征测量及实验室检查异常且有临床意义者;4)在首次服用试验用药品前2周内服用了任何处方及非处方药、维生素或中草药者;5)在首次服用试验用药品前参加过其他药物临床试验未满3个月者。
1.4 给药方案及血样采集
采用单中心、随机、开放、两周期、两序列、自身交叉对照的试验设计。将志愿者按照1:1的比例分配至T-R(受试制剂-参比制剂)和R-T(参比制剂-受试制剂)序列组,给药前至少禁食10 h,服药前、后1 h禁水,以(240±10)mL温水送服10 mg受试或参比制剂。空腹组在空腹状态下服药,餐后组在给药前30 min开始进食高脂餐,并于30 min内服用完毕。空腹和餐后组分别于给药4 h进食午餐,10 h后进食晚餐,餐食为营养食堂定制的标准餐,且保持两周期一致。
空腹、餐后组分别于给药前0 h和给药后0.25、0.5、0.75、1.、1.5、2、3、4、6、8、12、24、36、48 h采集4 mL静脉血至预冷的EDTA-K2抗凝管中,离心分离获得血浆并存储于-70 ℃冰箱保存。
1.5 血药浓度测定
采用高效液相-色谱串联质谱法测定人血浆中贝那普利及贝那普利拉的血药浓度,方法学验证和血药浓度测定由科文斯医药研发(上海)有限公司完成。检测方法经验证,在选择性、精密度、准确度、重现性等方面均符合要求,适用人血浆中贝那普利和贝那普利拉的定量测定。
高效液相色谱条件:色谱柱为Phenomenex,C18(50.0 mm×2.0 mm,5 μm),流速为0.60 mL·min-1。流动相A为含0.3%甲酸的水溶液;流动相B为含0.3%甲酸的乙腈:甲醇(50:50,v/v)溶液;进样量为5 μL,进样器温度为5 ℃。
质谱条件:电喷雾电离离子源,喷雾器电压5000 V;温度550 ℃;碰撞气压力8 psi(1 psi=6.895 kPa);喷雾气压力60 psi;气帘气体压力40 psi;辅助气压力65 psi;入口电势10 V;正离子方式检测;用于定量分析的离子反应分别为m/z 425.2→351.2(贝那普利)、m/z 397.2→351.2(贝那普利拉)、m/z 430.2→356.2(贝那普利-d5)、m/z 402.2→356.2(贝那普利拉-d5)。贝那普利和贝那普利拉的最低定量限和最低质控质量浓度均为1.00 ng·mL-1。
1.6 统计学方法
使用WinNonlin软件7.0版中的非房室模型,将贝那普利和贝那普利拉的主要药代动力学参数Cmax、AUC0-t、AUC0-∞经对数转换后再进行方差分析,方差分析模型中以给药序列、药物、周期作为固定效应,受试者顺序作为随机效应,计算主要指标的几何均值比的90%置信区间落在80.00%~125.00%,则判断2种制剂生物等效。对贝那普利及贝那普利拉Tmax进行非参数秩和检验,其余描述性与推断性统计分析使用SAS9.4软件。生物等效性评价基于原型药物贝那普利的分析。
2 结果
2.1 血药浓度-时间曲线
空腹组共筛选72人,最终入组30例健康志愿者;餐后组筛选76人,实际给药例数为30例。在空腹和餐后状态下单次口服10 mg盐酸贝那普利片的参比制剂和受试制剂后,盐酸贝那普利及代谢产物贝那普利拉的血药浓度-时间曲线,分别见图1及图2。
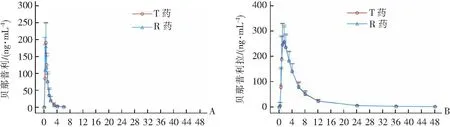
时间/h 时间/hA:贝那普利;B:贝那普利拉。图1 血药浓度-时间曲线(空腹组)
2.2 药代动力学参数
在空腹和餐后状态下,受试者口服10 mg盐酸贝那普利片后的药代动力学参数分别见表1—2。
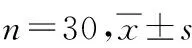
表1 空腹组口服盐酸贝那普利片后的药代动力学参数分析
表2 餐后组口服盐酸贝那普利片后的药代动力学参数分析
2.3 生物等效性评价
受试者在空腹及餐后条件下单次口服参比制剂或受试制剂后,血浆中贝那普利及代谢物贝那普利拉经对数转换的Cmax、AUC0-t和AUC0-∞几何均值比的90%置信区间,均在80.00%~125.00%等效区间内,Tmax在两种制剂间的差异无统计学意义(P>0.05),符合生物等效性评价标准。基于上述临床试验数据可以判断,深圳信立泰药业股份有限公司生产的受试制剂盐酸贝那普利片与原研的参比制剂盐酸贝那普利片(商品名:Cibacen®)生物等效。受试制剂相当于参比制剂的生物利用度F=AUC0-t(T)/AUC0-t(R),空腹组的F为(97.87±22.47)%,餐后组的F为(96.07±23.61)%。
2.4 安全性评价
空腹组不良事件发生率为23.3%(7/30);餐后组不良事件发生率为30.0%(9/30)。主要不良事件包括:甘油三酯升高、尿酸增高、腹痛、脉搏增快、血压降低、尿路感染、鼻衄、智齿冠周炎、干眼症、头晕。所有不良事件严重程度均为1级轻度,无严重不良事件发生。本研究结果表明,单次口服10 mg参比制剂和受试制剂后,受试者对该药具有良好的安全性和耐受性。
3 讨论
本研究数据显示:健康受试者口服10 mg贝那普利后,吸收迅速,空腹组约30 min达到贝那普利原型血药浓度的峰值Cmax,餐后组原型药约在1 h达到血药浓度的峰值Cmax,与文献[4-5]报道的一致。空腹组服药6 h后基本未测出原型药贝那普利的浓度,24 h基本未检测出活性代谢物贝那普利拉的浓度;而餐后组在48 h内持续能检测到原型药和活性代谢物的浓度,提示高脂饮食对贝那普利的吸收和代谢具有一定的影响。空腹组活性代谢物贝那普利拉的Cmax约出现在1.5 h,餐后组的贝那普利拉的Cmax约出现在2.5 h,均较原型药有所延长。根据国家药品监督管理局药品评审中心发布的《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》[6],有原型药和代谢物的制剂,一般推荐仅测定原型药物,因为原型药物的血药浓度-时间曲线比代谢产物能更灵敏地反映制剂间的差异。本研究同时检测了原型药物及活性代谢物的浓度,结果显示,两种制剂在健康中国人体内的药代动力学曲线的拟合度较高,提示该两种制剂在健康人体的吸收、分布、代谢、排泄等过程的差异较小,受试制剂与原研药具有生物等效性。
猜你喜欢
化工管理(2022年14期)2022-12-02
昆明医科大学学报(2021年6期)2021-07-31
中国现代医药杂志(2020年10期)2020-12-14
天津医科大学学报(2019年3期)2019-08-13
中国医院院长(2017年7期)2017-06-15
中国卫生标准管理(2015年5期)2016-01-14
中国继续医学教育(2015年4期)2016-01-07
中国药业(2014年17期)2014-05-26
中成药(2014年9期)2014-02-28
现代检验医学杂志(2014年1期)2014-02-06
