
失巢凋亡相关LncRNAs在肺腺癌中的预后价值及免疫浸润分析
2024-02-29 01:47:08李欣贺娟金山王若澜罗奇彪夏伟
肿瘤防治研究 2024年1期
李欣,贺娟,金山,王若澜,罗奇彪,夏伟
0 引言
肺癌是第二常见的癌症,也是全球癌症相关死亡的主要原因[1]。根据2020年肺癌发病率和死亡率的统计,新发肺癌患者220万例,新增死亡病例180万例[2]。肺腺癌(lung adenocarcinoma, LUAD)是非小细胞肺癌(non-small cell lung cancer, NSCLC)中最常见的亚型,大多数病例在诊断时已处于晚期并伴有转移[3]。虽然目前在临床诊断和治疗上取得了很大进步,但LUAD患者的5年生存率仍不理想[4]。因此,需要识别新型有效的生物标志物或风险分层方法,有助于LUAD患者的临床管理。
长链非编码RNA(long non-coding RNAs,lncRNAs)是长度超过200 nt,不编码蛋白质的线性RNA分子[5]。LncRNAs通过在转录、翻译和翻译后水平控制基因表达来调节不同的生物学功能[6]。其异常表达发生在各种恶性肿瘤的发生和发展过程中,许多lncRNAs已被证明是癌症诊断和治疗的潜在生物标志物和靶标[7-8]。目前,越来越多的证据表明lncRNAs在癌症中调控失巢凋亡和锚定非依赖性细胞生长发挥关键作用[9]。失巢凋亡(anoikis)是一种特殊类型的细胞死亡形式,对肿瘤细胞脱离细胞外基质(extracellular matrix, ECM)后的存活至关重要[10]。然而,关于失巢凋亡相关的lncRNAs(arlncRNAs)在LUAD中的预后以及肿瘤免疫微环境(tumor immune microenvironment, TIME)的研究还不清楚。
本研究基于生物信息学方法,通过arlncRNAs建立一个预后风险评分模型,进一步探讨该风险评分分型下患者肿瘤微环境的差异以及对化疗药物的敏感度分析,为LUAD患者的个性化治疗提供帮助。
1 资料与方法
1.1 数据来源
从TCGA数据库(https://portal.gdc.cancer.gov/)下载LUAD的转录组表达数据及相关临床信息,利用R语言对转录组数据进行整理后得到513例LUAD样本和58例正常组织样本测序数据。从GeneCards(https://www.genecards.org/)和Harmonizome(https://maayanlab.cloud/Harmonizome/)数据库获取了失巢凋亡相关的基因(ANRGs),其GeneCards数据库只纳入相关评分>1.0的基因,最后筛选得到360个。
1.2 失巢凋亡相关lncRNAs差异分析和加权基因共表达网络分析
从TCGA-LUAD的RNA表达谱中分离出mRNAs和lncRNAs的表达谱,在mRNAs表达谱中提取ANRGs的表达矩阵。对失巢凋亡相关mRNAs和lncRNAs进行共表达分析,采用Pearson相关性分析,以相关系数|R|>0.4和P<0.001为过滤标准,获得arlncRNAs。使用“DESeq2”包对arlncRNAs进行差异分析,以|log2FC|>1.0,Padj<0.05为筛选标准。然后,通过加权基因共表达网络分析(WGCNA)找到与肺腺癌发生密切相关的关键arlncRNAs。对上述两种分析方法的结果取交集后,筛选出LUAD发病机制中关键的arlncRNAs。
1.3 预后风险模型的建立与验证
首先,排除生存信息不完整且生存时间小于30天的样本,共获得490例。将TCGA-LUAD数据集中490例样本按7:3的比例随机分为训练集和测试集。对训练集进行单因素Cox分析和Lasso回归分析,最后通过多因素Cox分析确定建立风险模型的关键基因。通过以下公式计算各样本的风险评分:
按风险评分的中位数将样本分为低风险组和高风险组,通过Kaplan-Meier(K-M)生存分析和受试者工作特征曲线(ROC)来验证风险模型的预后预测能力。
1.4 列线图的构建和验证
为方便临床预测LUAD患者的预后,使用“rms”R包构建风险模型与其他临床因素1年、3年、5年的生存率列线图,根据校准曲线和一致性指数(C指数)评估列线图的预测能力,决策曲线分析法(DCA)评估列线图的临床净效益。
1.5 基因集富集分析
基于基因集富集分析(GSEA)方法对高低风险组之间差异表达的基因组进行富集分析,以探索两组之间的功能差异。从MSigDB数据库中下载c2.cp.kegg.v7.5.1.symbols.gmt作为参考基因集,富集的结果以P<0.05进行筛选。
1.6 肿瘤免疫微环境和免疫检查点的分析
为了进一步分析高低风险组间的免疫浸润情况,采用了几种公认的方法来探讨风险评分与免疫微环境之间的相关性,包括QUANTISEQ、MCPOUNTER、EPIC、CIBERSORT-ABS、CIBERSORT。运用ESTIMATE算法,评估两个风险组的TIME差异。通过单样本基因集富集分析(single sample geneset enrichment analysis,ssGSEA)来比较高、低风险组之间的免疫细胞浸润和免疫相关功能或通路的差异。最后,从以往的研究中获得46个与免疫检查点相关的基因[11],通过“ggpubr”R包比较两个风险组之间的免疫检查点表达情况。
1.7 药物敏感性分析
通过癌症药物敏感性基因组学数据库(genomics of drug sensitivity in cancer, GDSC)分析198种抗肿瘤药物的细胞系敏感性半数抑制浓度(IC50)数据。使用“oncoPredict”R包估计高、低风险组中每位LUAD患者对抗肿瘤药物的临床反应。
1.8 聚类分析
基于7个预后模型基因的表达水平,通过“ConsensusClusterPlus”R包对490例LUAD样本进行共识聚类[12]。采用“Rtsne”、“umap”R包进行t分布随机邻域嵌入(t-SNE)和统一流形逼近与投影(UMAP)验证聚类的可靠性。对不同亚型进行(Kaplan-Meier)生存分析,结合风险评分,观察每个亚型的风险分布情况。
1.9 统计学方法
研究中绘图和统计分析均使用R软件(4.1.3)和相关的R软件包进行。采用ROC曲线和K-M生存分析预测模型的准确性,单因素和多因素Cox回归分析验证模型是否具有独立预后价值,使用Wilcoxon秩和检验分析高、低风险组间的差异,Spearman相关性分析风险评分与免疫浸润细胞之间的相关性。P<0.05为差异有统计学意义。
2 结果
2.1 LUAD中失巢凋亡相关lncRNAs的鉴定
从TCGA-LUAD转录组数据中识别出8 236个lncRNAs和17 344个mRNAs,360个ANRGs在mRNAs表达矩阵中映射到340个,通过Pearson相关性分析获得2 339个arlncRNAs。对其结果进行差异分析和WGCNA分析,分别获得了966个差异表达的arlncRNAs(DE-arlncRNAs)和9个共表达模块,选取其中与肿瘤样本表型相关性最强的两个模块(具体分析过程请扫描本文OSID码)。合并这两个模块的基因并与差异基因取交集,得到448个在LUAD中差异表达的关键arlncRNAs,见图1。

图1 差异表达的arlncRNAs与关键模块arlncRNAs的维恩图Figure 1 Venn diagram of differentially expressed arlncRNAs versus key module arlncRNAs
2.2 构建预后模型
在训练集中通过单因素Cox分析发现448个arlncRNAs有93个与生存相关(P<0.05),根据P<0.01的标准,筛选到30个arlncRNAs。为更好地预测LUAD患者的预后,对30个arlncRNAs进行Lasso回归分析确定了17个arlncRNAs,最后根据多因素Cox分析,筛选出7个arlncRNAs构建了预后模型。图2A展示了预后模型中7个arlncRNAs的风险比,与ANRGs构建了共表达网络图,见图2B。每个患者的风险评分计算如下:
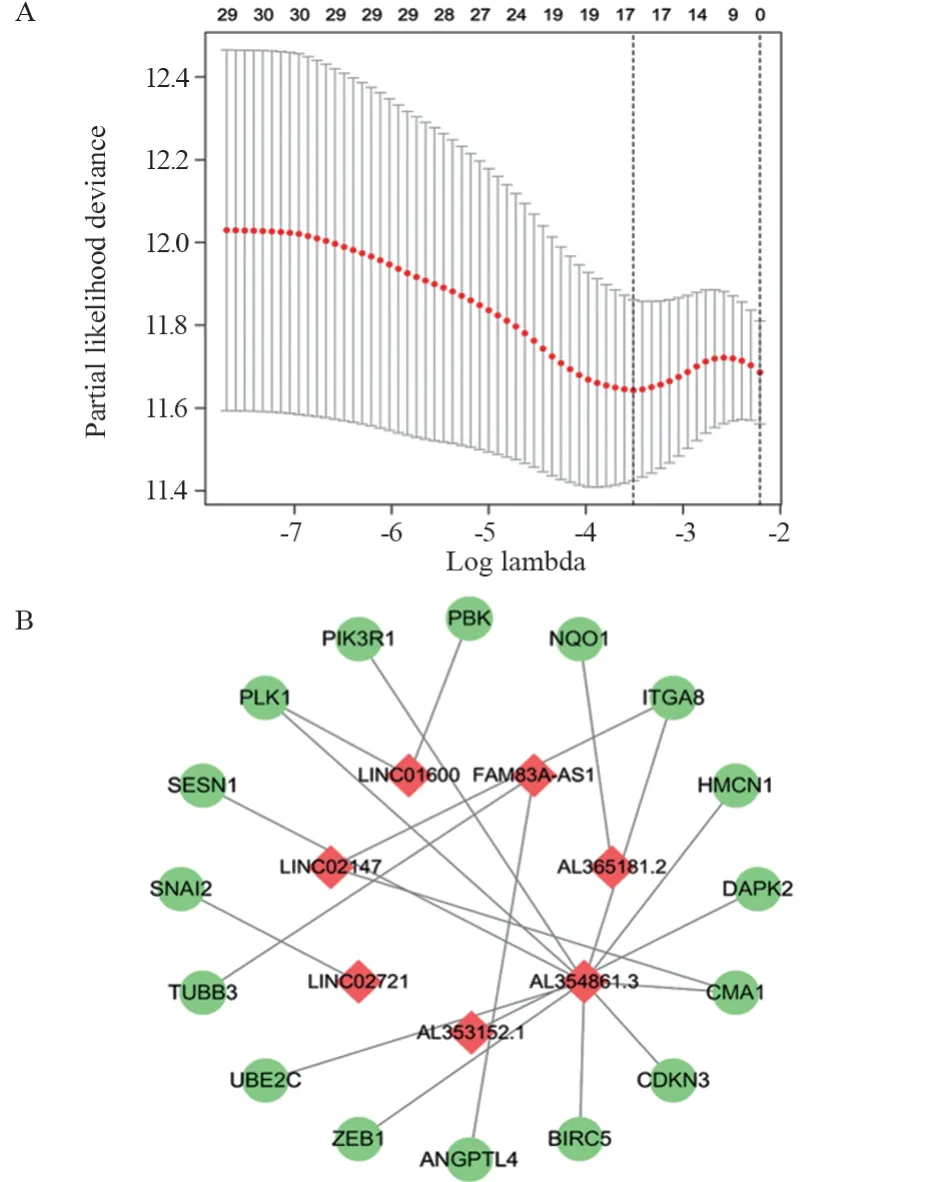
图2 基于预后相关的arlncRNAs构建风险模型Figure 2 Constructing a risk model based on prognosticrelated arlncRNAs
riskScore=`FAM83A-AS1`×(0.1603)+AL365181.2×(0.1277)+AL353152.1×(-0.4287)+AL354861.3×(-1.7937)+LINC02147×(-1.3384)+LINC02721×(2.1825)+LINC01600×(0.8042)。
2.3 预后模型的验证
通过组内验证来评估风险评分模型的可靠性,在训练集和测试集以及TCGA全集中,根据训练集风险评分的中位数将患者分为低风险组和高风险组。对患者的风险评分分布、生存状态和生存时间进行评估。结果显示,低风险组的总生存期(OS)远高于高风险组,见图3。亚组生存分析,进一步确定预后模型是否可以根据不同的临床特征预测患者的OS,按照年龄(≤65或>65)、性别(男性或女性)、临床分期(StageⅠ~Ⅱ或StageⅢ~Ⅳ)、T分期(T1~2或T3~4)、M分期(M0或M1)、N分期(N0或N1~3)进行亚组划分,观察到年龄、性别、M0分期、N分期、T分期、临床分期的临床特征,低风险组患者的OS高于高风险组患者(P<0.05),M1分期结果无统计学意义,见图4。综合上述结果说明除临床特征M1分期外,预后模型对不同临床特征分层的患者具有较好的预后预测能力。单因素和多因素Cox回归分析,结果表明该模型可作为一个独立的预后因素,见图5A~B。TCGA全集1年、3年和5年的ROC曲线下面积(AUC)分别为0.734、0.747和0.713,训练集和测试集的1年、3年、5年AUC也均大于0.7,见图5C~E。与其他临床特征比较,该模型具有较高的敏感度和特异性,见图5F。
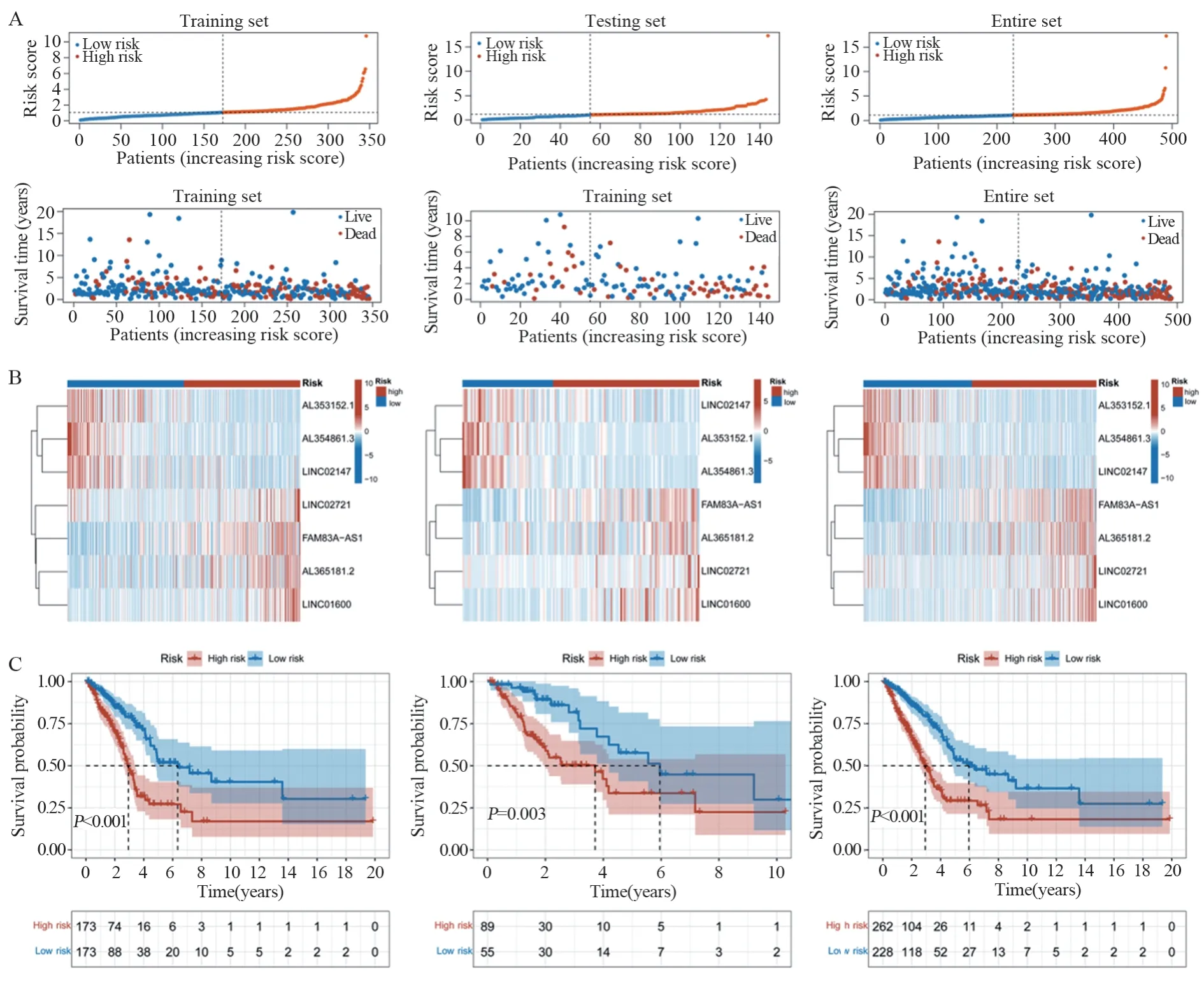
图3 训练集和两个验证集的风险评分分布、生存状态和生存时间的评估Figure 3 Evaluation of risk score distribution, survival status, and survival time for training set and two validation sets
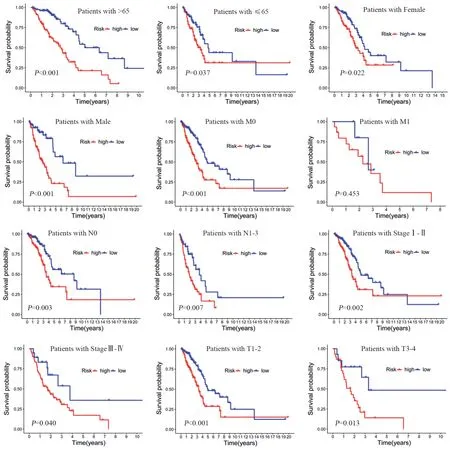
图4 整个TCGA数据集中高、低风险患者不同临床病理特征的生存分析Figure 4 Survival analysis of high- and low- risk patients in the entire TCGA dataset according to different clinicopathological characteristics
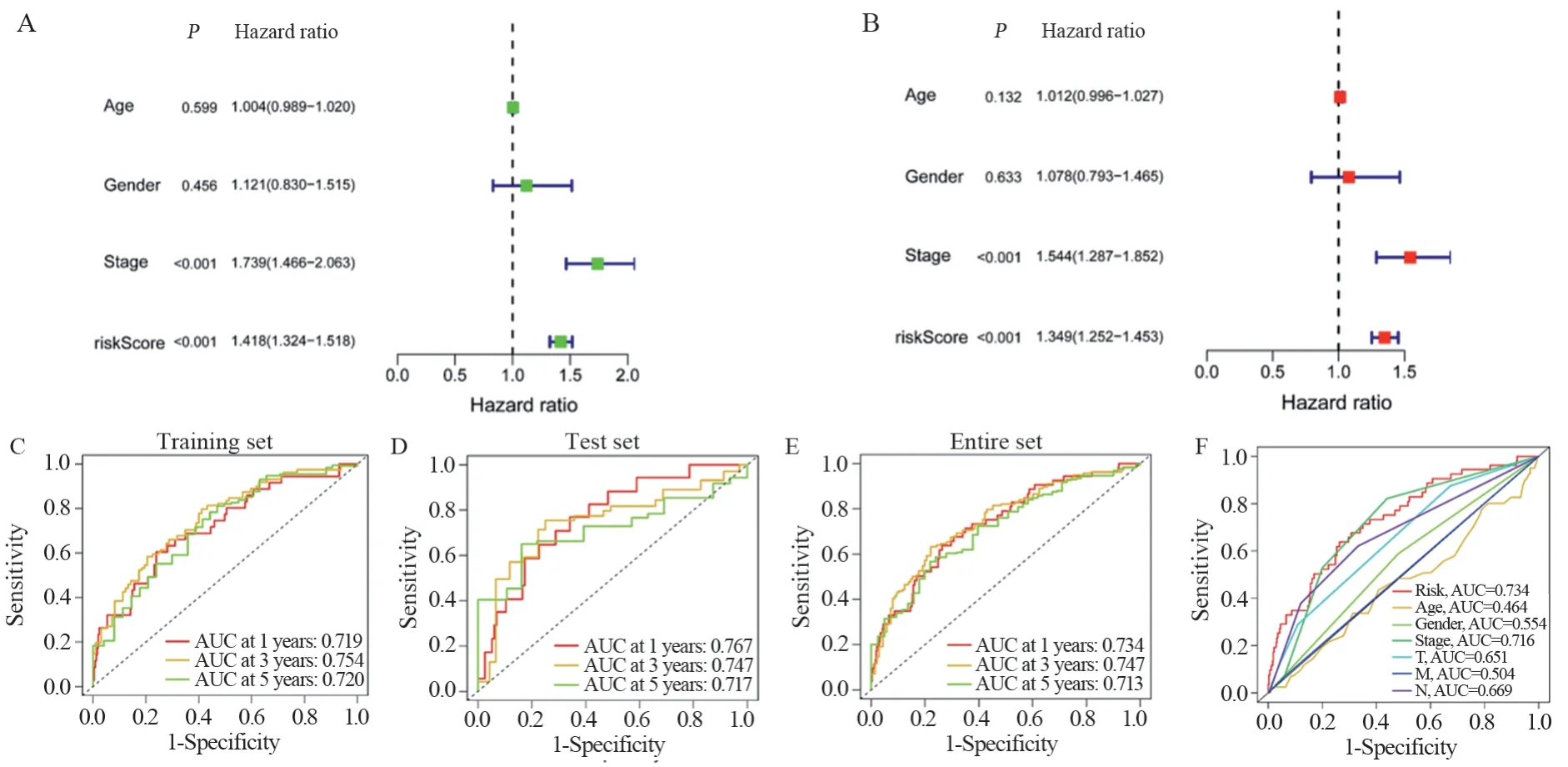
图5 风险模型性能的评估Figure 5 Evaluation of the risk model performance
2.4 列线图的构建
将图5B中显著的因素(stage、riskScore)用于构建列线图,见图6A。校准曲线显示了列线图在实际结果和预测结果之间表现出良好的一致性,见图6B,列线图的C指数为0.7017,表明列线图具有良好的预测价值。DCA结果显示,1、3、5年列线图相较于临床分期,列线图临床获益更大,见图6C。以上表明列线图的预测预后性能较好,有助于临床决策。
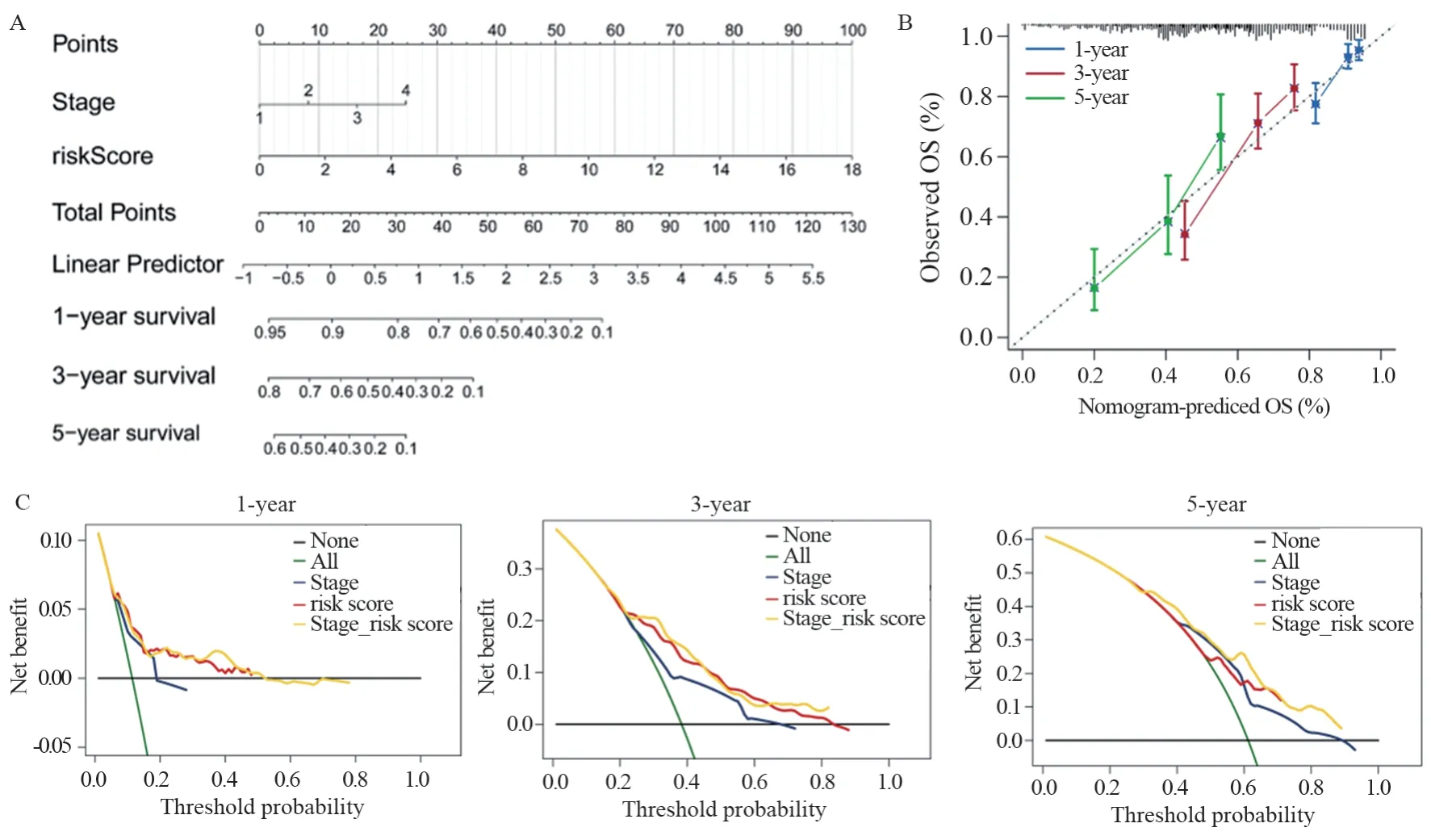
图6 列线图的构建和验证Figure 6 Construction and validation of nomogram
2.5 基于模型的高风险和低风险组的GSEA
为了研究不同风险组中可能参与的信号通路,GSEA结果见图7,发现高风险组中富集的通路主要与细胞周期、DNA复制、同源重组、剪接体、p53信号通路相关,低风险组与代谢相关通路、免疫相关通路相关联,包括花生四烯酸代谢、药物代谢-细胞色素P450、脂肪酸代谢、Fc epsilon RI信号通路。
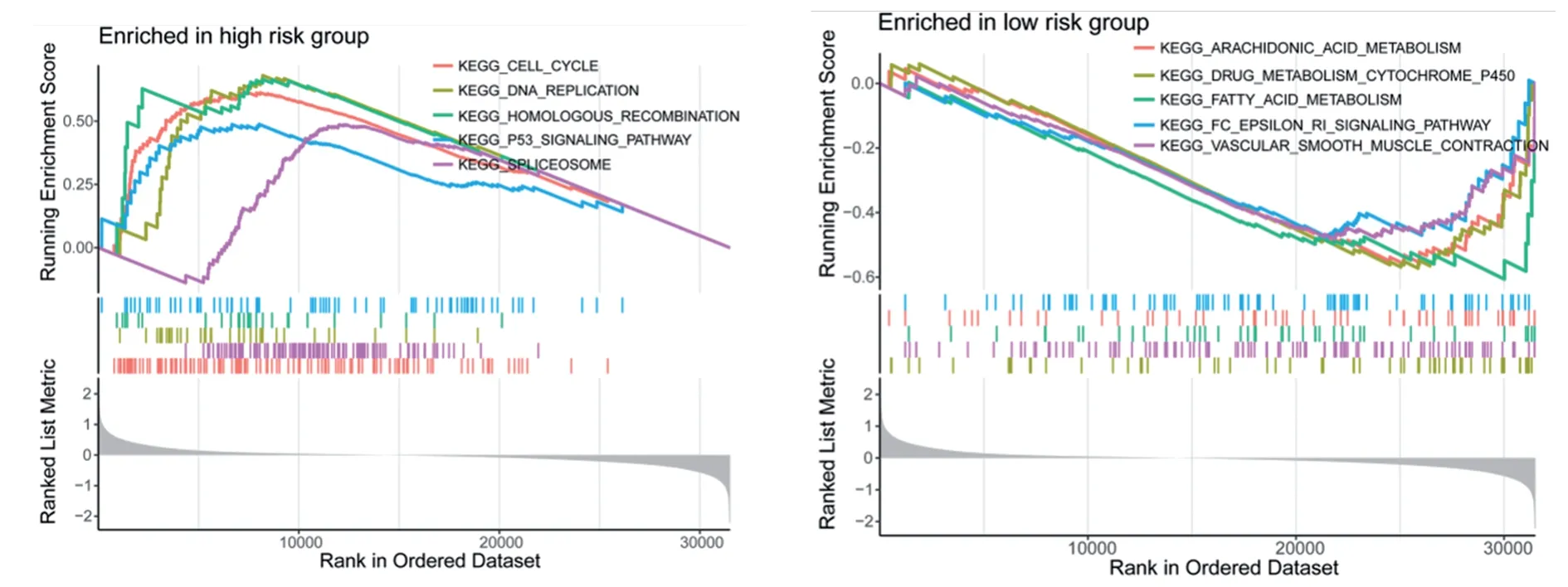
图7 高、低风险组之间的基因集富集分析Figure 7 Gene set enrichment analysis between high- and low-risk groups
2.6 不同风险组的免疫浸润分析及临床治疗探究
研究了风险评分与肿瘤浸润性免疫细胞的相关性,见图8A,不同平台上结果显示大多数免疫细胞与风险评分呈负相关。根据TIME评分,高风险组的免疫评分、基质评分、ESTIMATE评分更低,见图8B。ssGSEA分析显示,9个免疫细胞亚群的比例在低风险中都显著上升,免疫功能包括人类白细胞抗原(HLA)、Ⅰ型和Ⅱ型干扰素(IFN)反应在低风险组中得分更高,其他免疫功能没有显著性(P>0.05),见图8C~D。关于免疫检查点的表达差异分析,有27个在高低风险组间存在显著差异(P<0.05),见图8E,大部分在低风险组中表达上调。上述结果表明,低风险组的免疫浸润程度较高,这些患者的治疗可以选择更合适的免疫抑制剂。根据“oncoPredict”R包对两组的药物敏感性分析发现,高风险组对常用治疗LUAD药物如顺铂、多西他赛、紫杉醇和长春瑞滨更敏感(P<0.05),见图8F。因此风险评分可作为化疗敏感度的潜在预测指标,对LUAD患者的治疗有一定指导意义。
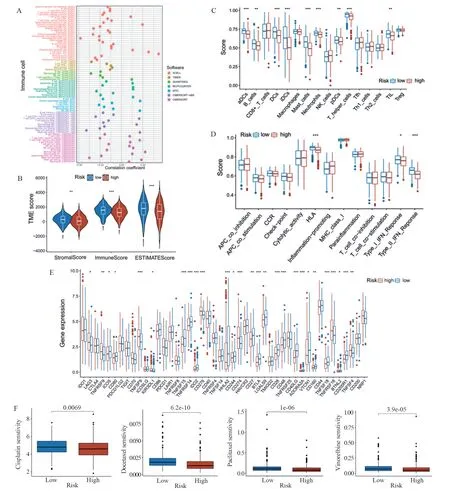
图8 高、低风险组患者的免疫浸润和药物敏感性分析Figure 8 Immune infiltration and drug sensitivity analysis of patients in high- and low-risk groups
2.7 基于7个模型基因的LUAD亚型分类
根据7个arlncRNAs的表达相似性,对LUAD患者进行亚型分类。当k=2时,确定了最优的聚类稳定性,将肺腺癌患者分为两个亚型C1(n=338)和C2(n=152)。使用t-SNE和UMAP验证了聚类的可靠性。分析两个亚型的预后以及在高低风险组的分布情况,发现与C2相比,C1预后好,且C1大部分分布在低风险组,几乎所有的C2分布在高风险组(分析过程图请扫描OSID码)。综上所述,这7个arlncRNAs在LUAD患者中表现出不同的表达模式,并可能影响患者的预后。
3 讨论
LUAD的发生和发展是一个涉及多种基因调控的复杂过程,通常在晚期被诊断为播散性转移性肿瘤[13-14]。失巢凋亡的抵抗认为是癌细胞转移的关键过程,使其成为有吸引力的癌症治疗靶点[15]。研究已经确定了几种因子,例如细胞黏附分子、生长蛋白、氧化应激、干细胞、自噬、非编码RNA和信号通路,用于调节失巢凋亡抵抗[16]。因此基于7个arlncRNAs构建了预后模型,旨在帮助预测LUAD患者的预后,并为LUAD患者开发新的治疗策略。
7个模型基因与OS显著相关,FAM83AAS1、AL365181.2、LINC02721、LINC01600是潜在的危险因素,AL353152.1、AL354861.3、LINC02147是潜在的保护因素。多项研究表明FAM83A-AS1在LUAD发展中发挥着重要作用[17-19],在LUAD中表达上调,促进LUAD的肿瘤发生,与较差的预后相关。LINC02147被确定是口腔粘膜下纤维化(oral submucous fibrosis, OSF)向口腔鳞状细胞癌(oral squamous cell carcinomas, OSCC)恶性进展相关的关键lncRNAs,低表达促进OSF恶性进展,并预测不良OSCC预后[20]。研究报道了LINC01600在胃肠道癌症中起致癌作用,高表达的患者生存期更短[21]。上述研究结果与本研究结果一致,FAM83A-AS1和LINC01600高表达是不利因素,LINC02147高表达是有利因素。在已发表的免疫相关的lncRNAs风险模型中,AL365181.2被选为标记基因,表明癌症中失巢凋亡和免疫调节之间可能存在联系[22]。而AL353152.1、AL354861.3、LINC02721的具体研究未见报道,其作用有待进一步阐明。因此,这些lncRNAs可能为肿瘤治疗提供新的预后生物标志物和治疗靶点。
GSEA的结果显示,高风险组中,细胞周期和DNA复制、p53信号通路等相关生物学途径被激活。低风险组中代谢途径和免疫相关途径被激活。由细胞周期失调和细胞周期蛋白依赖性激酶(cyclin-dependent kinases, CDK)激活导致的持续细胞增殖是肿瘤发生发展的重要标志[23]。基于这种风险模型,说明高风险人群癌症的进展更容易加速。探索风险评分与免疫微环境的关系表明低风险组中免疫细胞浸润水平较高。免疫功能方面,低风险组中HLA、Ⅰ型和Ⅱ型IFN反应显著增强。HLA与调节、监测和免疫反应密切相关,并在自身免疫性疾病、肿瘤免疫和生殖免疫中发挥重要作用[24]。Ⅰ型IFN和Ⅱ型IFN长期以来被认为是关键的抗病毒和抗微生物分子[25]。Ⅰ型IFN除了直接的抗病毒作用,还调节适应性免疫反应,能通过阻断细胞周期进程和诱导细胞凋亡直接作用于肿瘤或通过引发免疫细胞来促进肿瘤清除和防止转移[26-28]。Ⅱ型IFN主要由免疫细胞(活化的T细胞、自然杀伤细胞和巨噬细胞)产生,在抗肿瘤增殖和细胞免疫中发挥重要作用[29]。然而,研究报道乳腺癌、黑色素瘤和胃肠癌患者的淋巴细胞IFN信号缺陷,可能代表了一种常见的免疫功能障碍的癌症相关机制[30]。因此推测高风险组患者Ⅰ型IFN应答和Ⅱ型IFN应答的低表达可能是由于肿瘤的免疫缺陷。
免疫检查点的分析表明不同风险组可能对免疫治疗的反应有所不同,同时为LUAD免疫治疗提供了新的想法和选择。药物敏感性分析发现,高风险组患者对常用化疗药物顺铂、多西他赛、紫杉醇、长春瑞滨更敏感。上述GSEA发现细胞周期和DNA修复等相关途径在高风险组中显著富集。因此,这些药物可能通过不同的机制来干扰细胞周期。
此外,共识聚类分析表明了这7个基因不仅可作为预测LUAD患者总体生存的候选分子标志物,还可用于患者聚类,并为具有不同分子性质的患者确定最佳候选药物。然而,本研究也存在一些局限性和缺点。首先,需要包括来自不同队列的数据用于外部验证,其次研究基于公共数据库的回顾性分析,某些lncRNAs在LUAD中的机制尚不清楚。因此,在接下来的工作中,我们将收集和扩充临床样本来验证该模型的准确性和稳健性,通过更多的前瞻性研究来探索特征基因的临床价值。
利益冲突声明:
所有作者均声明不存在利益冲突。
猜你喜欢
现代临床医学(2023年4期)2023-09-26 09:40:38
核安全(2022年2期)2022-05-05 06:55:32
疯狂英语·新读写(2021年10期)2021-12-07 02:41:30
中国民间疗法(2021年18期)2021-11-02 08:20:26
供水技术(2020年6期)2020-03-17 08:18:36
奥秘(2019年8期)2019-08-28 01:47:05
商周刊(2017年7期)2017-08-22 03:36:21
东北史地(学问)(2016年6期)2016-12-14 02:03:20
外语教学理论与实践(2016年1期)2016-06-11 05:51:40
小猕猴智力画刊(2016年6期)2016-05-14 09:21:40
