
郁金香素A拼接双螺环恶嗪啉-氧化吲哚的合成
2024-02-28 01:40石庆辉王慧娟姜维艳刘雄利邓国栋
合成化学 2024年2期
任 娜, 石庆辉, 王慧娟, 姜维艳,2*, 刘雄利, 邓国栋*
(1. 贵州大学 西南药食两用资源开发利用技术国家地方联合工程研究中心,贵州 贵阳 550025; 2. 贵州装备制造职业学院,贵州 贵阳 550025)
郁金香素A(Tulipalin A)属于倍半萜内酯家族,该骨架存在于许多具有生物学意义的天然产物中[1-2]。 1,2-恶嗪烷也是一个优势的杂环骨架,其存在于大量具有生物活性的天然生物碱和与药物相关的化合物中,如Pretrichodermamide A和Pretrichodermamide C, Gliovirin和Penicisulfuranols A~F(图1)[3-6]。哈佛大学SCHREIBER教授于2000年提出“多样性导向合成”(DOS)概念[7-9],其以一种“高通量”的方式产生“类天然产物”的衍生物库。因此,开发一种有效的方法,用容易获得的原料构建郁金香素A拼接的1,2-恶嗪烷骨架衍生物,对于发现新的生物活性分子具有重要的研究意义。

图1 含有1,2-恶嗪烷骨架的天然产物分子Figure 1 Natural product molecules containing 1,2-oxazine skeleton
螺环氧化吲哚的结构复杂性和立体中心的丰富性极大地影响了其生物活性[10-13],因此,开发一种有效而直接的方法提高螺环氧化吲哚的结构多样性,对于进一步生物测试具有重要研究意义,但同时也具有挑战性。通过文献调研,将郁金香素A和恶嗪啉拼接到螺环氧化吲哚骨架上,可以提高螺环氧化吲哚的结构多样性,目前该合成方法未见文献报道。
本文以靛红和脯氨酸为起始原料,先与郁金香素A发生[3+2]环加成反应[14],生成环加成中间体3,然后中间体3中的氮原子在氧化剂间氯过氧苯甲酸(m-CPBA)的作用下发生N-氧化反应[15-17],得到不稳定的中间体3′,随后中间体3′发生分子内1,2-迁移和扩环反应,得到8个新型郁金香素A拼接恶嗪啉双螺环氧化吲哚(4a~4h,图2),总产率50%~62%,dr>20 ∶1。化合物结构经1H NMR,13C NMR和HR-MS(ESI-TOF)表征,其中,化合物4g的相对构型通过单晶X-射线衍射进行了进一步确定。该类新型拼接衍生物可为生物活性测试提供新化合物筛选[18-20]。
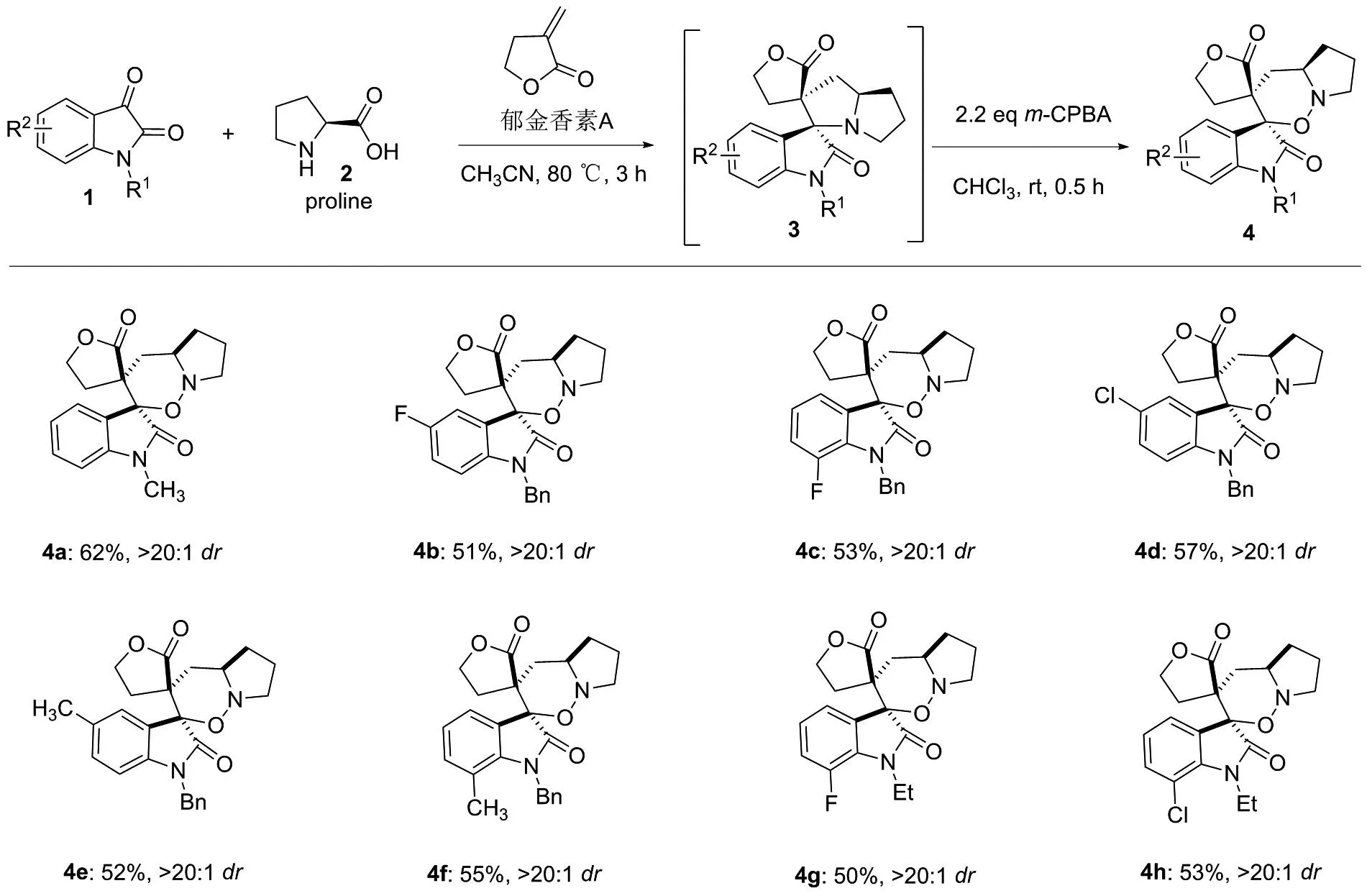
图2 郁金香素A拼接恶嗪啉螺环氧化吲哚的合成路线Figure 2 Synthesis route of tulipalin A-based bispiro[oxazinane-oxindole] hybrids
1 实验部分
1.1 仪器与试剂
WRS-1B型数字熔点仪; Bruker-400 MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标); MicroTMQ-TOF型高分辨质谱仪。
所用试剂均为分析纯。
1.2 4a~4h的合成(以4a为例)
在装有磁力搅拌子的密封管中将0.3 mmol靛红1、 0.2 mmol郁金香素A和0.5 mmol脯氨酸2加入2.0 mL的CH3CN中,在80 ℃下搅拌3 h,然后使用洗脱剂(石油醚 ∶乙酸乙酯=5 ∶1,V∶V)通过快速色谱法纯化,得到所需中间体3。将0.2 mmol中间体3, 2.2 eqm-CPBA加入2.0 mL的CHCl3中,室温下搅拌30 min,然后使用洗脱剂(甲醇 ∶乙酸乙酯=1∶20,V∶V)通过快速色谱法纯化,得到所需目标产物4a,淡黄色固体,m.p.256.3~257.5 ℃,总产率62%,dr>20∶1;1H NMRδ: 0.97~1.01(m, 1H), 1.69~1.74(m, 1H), 1.80~1.86(m, 2H), 1.94~1.99(m, 1H), 2.12(d,J=12.8 Hz, 1H), 2.25~2.33(m, 1H), 2.50~2.53(m, 1H), 2.61~2.68(m, 1H), 2.78~2.80(m, 1H), 3.12~3.14(m, 1H), 3.17(s, 3H), 3.42~3.44(m, 1H), 4.02~4.08(m, 1H), 4.28~4.34(m, 2H), 7.05(d,J=7.6 Hz, 1H), 7.10~7.14(m, 1H), 7.40~7.44(m, 1H), 7.96(d,J=7.2 Hz, 1H);13C NMRδ: 14.0, 19.1, 25.6, 26.7, 30.5, 52.4, 60.7, 65.5, 66.3, 80.9, 109.0, 122.4, 129.1, 130.1, 132.0, 143.3, 171.6, 176.3; HR-MS(ESI-TOF)m/z: calcd for C18H20N2NaO4{[M+Na]+}351.1315, found 351.1318。
用类似的方法合成4b~4h。
4b:淡黄色固体,m.p.250.2~251.1 ℃,总产率51%,dr>20∶1;1H NMRδ: 1.56~1.59(m, 1H), 1.73~1.81 (m, 2H), 1.85~1.92(m, 2H), 2.04~2.12(m, 1H), 2.51~2.57(m, 1H), 2.63~2.74(m, 2H), 3.14~3.19(m, 1H), 3.63(s, 1H), 4.02~4.08(m, 1H), 4.12~4.18(m, 1H), 4.55(d,J=16.0 Hz, 1H), 4.95(d,J=16.0 Hz, 1H), 6.46~6.49(m, 1H), 6.80~6.85(m, 1H), 7.11~7.25(m, 5H), 7.69~7.72(m, 1H);13C NMRδ: 16.7, 24.1, 29.3, 35.7, 42.5, 46.4, 51.0, 58.8, 64.7, 79.5, 108.0(d,JCF=8.5 Hz), 114.4(d,JCF=23.4 Hz), 116.5(d,JCF=26.1 Hz), 125.2, 126.2, 127.3, 133.1, 136.6, 156.8(d,JCF=238.7 Hz), 170.4, 174.5; HR-MS(ESI-TOF)m/z: calcd for C24H23FN2NaO4{[M+Na]+}445.1534, found 445.1536。
4c:淡黄色固体,m.p.245.6~246.1 ℃,总产率53%,dr>20 ∶1;1H NMRδ: 1.63(s, 1H), 1.73~1.91(m, 4H), 2.01~2.09(m, 1H), 2.53~2.59(m, 1H), 2.64~2.79(m, 2H), 3.14~3.19(m, 1H), 3.53(s, 1H), 3.91~3.98(m, 1H), 4.08~4.14(m, 1H), 4.89~4.98(m, 2H), 6.91~6.97(m, 2H), 7.16~7.24(m, 5H), 7.74(s, 1H);13C NMRδ: 17.0, 24.2, 29.5, 35.2, 44.0, 46.0, 51.3, 58.9, 64.4, 79.0, 116.4(d,JCF=19.8 Hz), 121.4(d,JCF=7.7 Hz), 124.0, 125.5, 125.9, 126.9, 127.4(d,JCF=9.6 Hz), 128.0, 134.5, 145.6(d,JCF=242.5 Hz), 170.2, 174.5; HR-MS(ESI-TOF)m/z: calcd for C24H23FN2NaO4{[M+Na]+}445.1534, found 445.1537。
4d:淡黄色固体,m.p.251.4~252.8 ℃,总产率57%,dr>20∶1;1H NMRδ: 1.58~1.65(m, 1H), 1.72~1.83(m, 2H), 1.89~1.96(m, 1H), 2.09(d,J=11.6 Hz, 1H), 2.24~2.32(m, 1H), 2.37~2.43(m, 1H), 2.56~2.63(m, 1H), 2.73~2.75(m, 1H), 3.11~3.15(m, 1H), 3.46~3.48(m, 1H), 3.93~4.00(m, 1H), 4.24~4.30(m, 1H), 4.78(d,J=15.6 Hz, 1H), 4.98(d,J=15.6 Hz, 1H), 6.91(d,J=8.4 Hz, 1H), 7.22~7.35(m, 6H), 7.90(s, 1H);13C NMRδ: 14.4, 18.2, 25.5, 30.0, 43.5, 47.9, 52.6, 60.7, 66.5, 80.9, 111.2, 126.5, 127.4, 128.1, 129.2, 129.9, 130.0, 135.8, 141.4, 171.4, 176.3; HR-MS(ESI-TOF)m/z: calcd for C24H23ClN2NaO4{[M+Na]+}461.1239, found 461.1232。
4e:淡黄色固体,m.p.265.2~266.1 ℃,总产率52%,dr>20 ∶1;1H NMRδ: 1.73~1.86(m, 4H), 2.01~2.09(m, 2H), 2.25(s, 3H), 2.54~2.60(m, 1H), 2.67~2.73(m, 1H), 2.80(s, 1H), 3.17~3.21(m, 1H), 3.41~3.47(m, 1H), 3.96~4.04(m, 1H), 4.09~4.14(m, 1H), 4.57(d,J=16.0 Hz, 1H), 4.93(d,J=16.0 Hz, 1H), 6.46(d,J=16.0 Hz, 1H), 6.92~6.94(m, 1H), 7.13~7.24(m, 5H), 7.68(s, 1H);13C NMRδ: 16.7, 19.0, 23.5, 29.1, 34.0, 41.5, 44.9, 51.1, 58.4, 63.5, 78.6, 106.5, 124.5, 125.1, 126.4, 127.9, 129.5, 132.8, 137.6, 169.9, 173.9; HR-MS(ESI-TOF)m/z: calcd for C25H26N2NaO4{[M+Na]+}441.1785, found 441.1789。
4f:淡黄色固体,m.p.240.1~241.0 ℃,总产率55%,dr>20 ∶1;1H NMRδ: 1.67~1.87(m, 4H), 1.94~196(m, 1H), 1.20~2.08(m, 1H), 2.12(s, 3H), 2.58~2.77(m, 3H), 3.15~3.20(m, 1H), 3.50~3.53(m, 1H), 3.88~3.94(m, 1H), 4.06~4.11(m, 1H), 4.94(d,J=16.8 Hz, 1H), 5.15(d,J=16.8 Hz, 1H), 6.88~6.94(m, 2H), 7.05(d,J=7.2 Hz, 2H), 7.12~7.16(m, 1H), 7.20~7.24(m, 2H), 7.87(d,J=6.0 Hz, 1H);13C NMRδ: 17.5, 24.5, 30.0, 35.3, 43.9, 46.2, 51.6, 59.1, 64.6, 78.5, 118.1, 121.0, 123.9, 125.7, 126.1, 126.3, 127.4, 132.7, 135.5, 139.0, 171.9, 175.0; HR-MS(ESI-TOF)m/z: calcd for C25H26N2NaO4{[M+Na]+}441.1785, found 441.1780。
4g:淡黄色固体,m.p.271.3~271.9 ℃,总产率50%,dr>20 ∶1;1H NMRδ: 1.16~1.19(m, 3H), 1.59(s, 1H), 1.74~1.80(m, 2H), 1.83~1.91(m, 2H), 2.02~2.10(m, 1H), 2.50~2.56(m, 1H), 2.63~2.73(m, 2H), 3.13~3.18(m, 1H), 3.54(s, 1H), 3.71~3.78(m, 1H), 3.82~3.88(m, 1H), 4.02~4.08(m, 1H), 4.14~4.19(m, 1H), 6.91~7.03(m, 2H), 7.75(d,J=7.2 Hz, 1H);13C NMRδ: 13.8, 18.5, 25.8, 31.0, 37.2, 47.9, 52.8, 53.5, 60.5, 66.3, 80.8, 118.0(d,JCF=19.4 Hz), 122.8(d,JCF=6.3 Hz), 125.7, 128.9(d,JCF=8.4 Hz), 130.1, 147.6(d,JCF=242.1 Hz), 171.4, 176.2; HR-MS(ESI-TOF)m/z: calcd for C19H21FN2NaO4{[M+Na]+}383.1378, found 383.1382。
4h:淡黄色固体,m.p.244.6~245.5 ℃,总产率53%,dr>20 ∶1;1H NMRδ: 1.19~1.22(m, 3H), 1.63(s, 1H), 1.74~1.92(m, 4H), 2.01~2.09(m, 1H), 2.50~2.56(m, 1H), 2.64~2.74(m, 2H), 3.14~3.19(m, 1H), 3.51~3.52(m, 1H), 3.99~4.19(m, 4H), 6.90~6.94(m, 1H), 7.19~7.21(m, 1H), 7.91(d,J=7.2 Hz, 1H);13C NMRδ: 13.2, 17.3, 24.6, 30.0, 35.7, 46.5, 51.7, 59.2, 64.9, 78.6, 113.8, 121.8, 126.8, 128.8, 131.2, 137.0, 171.0, 174.8; HR-MS(ESI-TOF)m/z: calcd for C19H21ClN2NaO4{[M+Na]+}399.1082, found 399.1086。
2 结果与讨论
2.1 底物拓展分析
由于第1步环加成反应后,反应溶剂里含有未反应完的原料和杂质,如果不分离中间体3,直接进行第2步的氧化重排反应,最终产物4分离纯化将变得困难,所以本实验采取分离中间体再进行氧化重排反应策略。通过对底物进行拓展可以发现(图2),该类反应对底物靛红1的取代基没有影响,底物靛红1不管是给电子取代(1e和1f)还是吸电子取代(1b~1d,1g和1h),都能以较好的产率(总产率高达62%)和优异的非对映选择性(dr>20 ∶1)得到所需的产物4a~4h,表现出极好的立体选择性。
2.2 单晶分析
图3为化合物4g的单晶结构。由图3分析可知,化合物4g属Monoclinic晶系,P21/n空间群,晶胞参数a=1.48192(13) nm,b=0.83536(5) nm,c=1.49317(13) nm,α=90°,β=116.626(11)°,γ=90°。
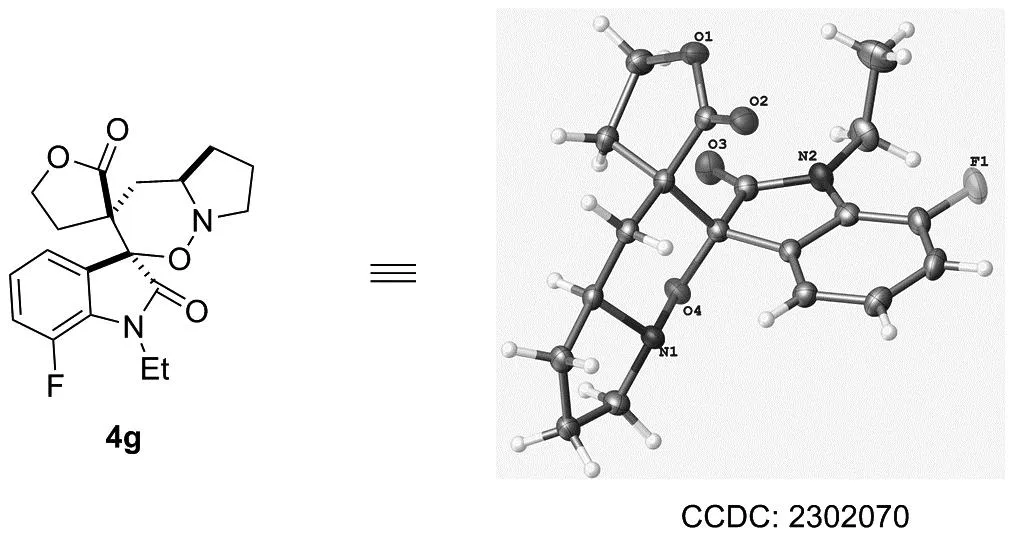
图3 化合物4g的单晶结构Figure 3 Single crystal structure of compound 4g
2.3 反应机理分析
该反应的可能机理如图4所示。靛红1和脯氨酸2通过脱羧过程产生甲亚胺叶立德A。所形成的叶立德A继续和底物郁金香素A发生[3+2]环加成反应,从而得到环加成中间体3。中间体3在m-CPBA(间氯过氧苯甲酸)的氧化下,发生N-氧化反应得到中间体3′,然而中间体3′不稳定,可能是由于中间体3′中3个相邻的四取代立体中心之间的排斥作用,从而导致N-氧化物基团中的氧原子通过1,2-迁移过程攻击氧化吲哚的C3螺环中心,扩环得到目标产物郁金香素A拼接恶嗪啉双螺环氧化吲哚4。
本文以靛红和脯氨酸为起始原料,先与郁金香素A发生[3+2]环加成反应,生成环加成中间体3,然后中间体3中的氮原子在氧化剂m-CPBA的作用下发生N-氧化反应,得到不稳定的中间体3′,随后中间体3′发生分子内1,2-迁移和扩环反应,得到8个新型目标产物郁金香素A拼接恶嗪啉螺环氧化吲哚(4a~4h),总产率50%~62%,dr>20 ∶1,其结构经1H NMR,13C NMR和HR-MS(ESI-TOF)表征。化合物4g的相对构型通过单晶X-射线衍射进一步确定。该类新型拼接衍生物今后可为生物活性测试提供新化合物基础,对于后续发现生活活性先导化合物具有重要的研究意义。
猜你喜欢
今日农业(2022年13期)2022-11-10
今日农业(2021年15期)2021-11-26
中老年保健(2021年4期)2021-08-22
今日农业(2020年17期)2020-10-27
时代英语·高一(2019年5期)2019-09-03
扬子江(2019年1期)2019-03-08
快乐语文(2016年15期)2016-11-07
中国中医药信息杂志(2016年3期)2016-03-02
好孩子画报(2014年11期)2014-11-12
少儿科学周刊·少年版(2014年1期)2014-04-11
