
超高效液相色谱-三重四极杆质谱法测定化妆品中8种替丁类禁用药物
2024-02-27 15:16:52张秋炎梁维维罗辉泰廖均涛吴惠勤
分析测试学报 2024年2期
张秋炎,梁维维,罗辉泰,廖均涛,黄 芳,吴惠勤
(广东省科学院测试分析研究所(中国广州分析测试中心),广东省化学测量与应急检测技术重点实验室,广东省中药质量安全工程技术研究中心,广东 广州 510070)
化妆品产业高质量发展的同时,其安全问题也日益引起消费者的重视。药物非法添加行为屡禁不止,给消费者的使用带来潜在风险[1]。以西咪替丁、雷尼替丁为代表的替丁类药物是组胺H2受体拮抗剂,能有效降低胃酸,多用于治疗消化性溃疡,如十二指肠溃疡和胃溃疡等。此外,替丁类药物具有阻断双氢睾酮与受体结合的作用,可抑制皮脂分泌,故常单独或联合应用治疗带状疱疹、荨麻疹、过敏性紫癜、湿疹、痤疮等皮肤病[2-3]。西咪替丁已被国家药品监督管理局列为化妆品中主要检测物质之一,并公布了其检测方法,但该方法仅对西咪替丁进行分析,并未针对其他替丁类药物[4]。不法企业为了规避政府监管,可能添加与西咪替丁功能相似的其他替丁类药物替代西咪替丁。常见替丁类药物包括雷尼替丁、罗沙替丁等8种,化学结构式如图1所示。目前针对化妆品中替丁类药物的测定方法主要有液相色谱法[5-6]、液相色谱-质谱联用法[7-9]等,但上述方法涉及的替丁类化合物最多仅3 种,未包括常见的替丁类药物。因此,建立涵盖种类更齐全的替丁类化合物检测方法尤为迫切,对保障化妆品质量安全具有重要的现实意义。
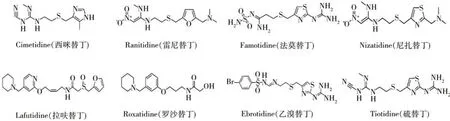
图1 8种替丁类药物的化学结构式Fig.1 Chemical structures of eight tidine drugs
本文首次建立了可同时测定5种不同类型化妆品基质(包括水剂类、膏霜类、乳液类、凝胶类和粉剂类)中8 种替丁类药物的超高效液相色谱-串联质谱法(UHPLC-MS/MS)。本方法简单高效、易操作、灵敏度高、可行性强,目前已成功应用于分析检测工作中。本法可作为政府部门有效的技术支持和补充方案,为化妆品中非法添加药物监测提供科学依据,便于化妆品市场的监管。
1 实验部分
1.1 仪器、试剂与材料
Agilent 1290 Ⅱ UHPLC/6470A Triple Quard MS超高效液相色谱/串联四极杆质谱联用仪(美国Agilent公司);AS 3120 超声波发生器(天津奥特赛恩斯仪器有限公司);TP-114 电子天平(美国Sartorious 公司);XW-80A 快速混匀器(海门市麒麟医用仪器厂);1850R 高速冷冻离心机(湖南湘仪实验室仪器开发有限公司);YMC-Triart C18色谱柱(100 mm×2.1 mm,3.0 μm,日本株式会社YMC)。
乙酸、甲酸、乙酸铵及甲酸铵为LC-MS 级(美国Sigma 公司);乙腈(ACN)及甲醇(MeOH)为LCMS级(德国Merck公司);实验用水为二次蒸馏水,其余所用试剂为分析纯。
8 种对照品:西咪替丁(纯度99.8%)、雷尼替丁(纯度99.9%)、法莫替丁(纯度99.5%)、尼扎替丁(纯度99.6%)、拉呋替丁(纯度99.8%)及盐酸罗沙替丁醋酸酯(纯度100%)购于中国食品药品检定研究院;乙溴替丁及硫替丁(质量浓度100 μg/mL)购于天津阿尔塔科技有限公司。所用化妆品样品来源于市场购买或客户委托送检。
1.2 标准溶液配制
分别称取西咪替丁、雷尼替丁、法莫替丁、尼扎替丁、拉呋替丁、盐酸罗沙替丁醋酸酯约10.0 mg至10 mL容量瓶中,以甲醇作为溶剂超声定容至刻度,制得质量浓度约为1 000 μg/mL的单标标准储备液,转移至棕色储液瓶中,4 ℃保存。另分别量取乙溴替丁和硫替丁各1 mL 至10 mL 容量瓶中,用甲醇定容至刻度,涡旋均匀得到10 μg/mL的单标标准储备液,转移至棕色储液瓶中,4 ℃保存。
1.3 样品处理
称取样品约0.2 g(精确至0.01 g)于10 mL具塞比色管中,加入2 mL水,在旋涡混合器上高速振荡30 s 后用乙腈定容至刻度,涡旋振荡1 min 后超声提取15 min,待提取液静置至室温,4 000 r/min 下离心10 min。取上清液经0.22 μm滤膜过滤,滤液上机分析。必要时用适量80%乙腈稀释。
1.4 仪器测定条件
1.4.1 液相色谱条件色谱柱:YMC-Triart C18(100 mm×2.1 mm,3.0 μm);流动相:A相为0.2%(体积分数,下同)甲酸溶液,B 相为乙腈;流速:0.35 mL/min;梯度洗脱程序:0~1.00 min,5% B;1.00~6.00 min,5%~20% B,6.00~9.00 min,20%~65% B;9.00~9.01 min,65%~95% B;9.01~10.50 min,95% B;10.50~10.51 min,95%~5% B;10.51~12.00 min,5% B。进样量:0.5 μL;柱温:30 ℃;分析时间:12.0 min。
1.4.2 质谱条件用喷射流电喷雾离子源(AJS ESI)在正离子模式下检测;采集方式为多反应监测(MRM);干燥气流速为10 L/min;干燥气温度为325 ℃;鞘气流速为11 L/min;鞘气温度为350 ℃;雾化气压力为310 kPa;毛细管入口端电压为4 000 V;喷嘴电压为500 V。8 种替丁类药物的化合物信息及质谱采集参数见表1。
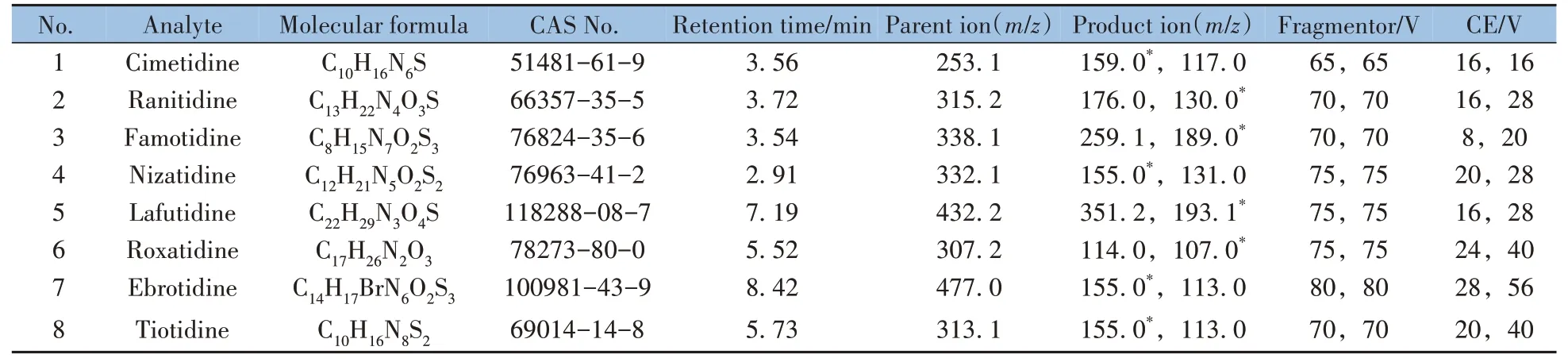
表1 8种替丁类药物的分子式、CAS号、保留时间及质谱采集参数Table 1 Molecular formula,CAS registration number(CAS No.) ,retention time and MS parameters of eight tidine drugs
2 结果与讨论
2.1 质谱条件的优化
分别配制1 mg/L 的8 种替丁类药物单标标准溶液,在正、负离子模式下进行一级全扫描分析,发现8 种替丁类药物在[M+H]+加合方式下响应最高。原因可能是8 种替丁类药物均为含氮化合物,氮原子的孤对电子易接受质子,形成[M+H]+的准分子离子峰。选择仪器自带的Optimizer软件对各化合物的特征子离子、碰撞能量和碎裂电压等质谱参数进行优化以达到欧盟2002/657/EC 关于4 个识别点的要求[10]。8种替丁类药物的质谱采集参数见表1。
2.2 色谱条件的优化
2.2.1 色谱柱的选择比较了5 种不同型号色谱柱YMC-Triart C18(100 mm×2.1 mm,3.0 μm)(A)、ZORBAX RRHD Eclipse Plus C18(100 mm×3.0 mm,1.8 μm)(B)、ZORBAX SB-C8RRHD(100 mm×3.0 mm,1.8 μm)(C)、Poroshell 120 PFP(100 mm×2.1 mm,2.7 μm)(D)和CORTECS UPLC@T3(150 mm×2.1 mm,1.6 μm)(E)的分离效果。结果表明:8 种替丁类药物在D 柱上的保留差,出峰较早,分离效果较差;C 柱上雷尼替丁和尼扎替丁存在色谱峰过宽或分叉等情况;8 种替丁类药物在A 柱、B 柱和E柱上有较好的保留,且分离效果理想,雷尼替丁和尼扎替丁的峰形均无拖尾现象,其他6 种替丁类药物的峰形良好;此外8种替丁类药物在A 柱上的响应最高,B 柱次之。综合比较,最终选择A 柱YMCTriart C18(100 mm×2.1 mm,3.0 μm)作为色谱分离柱。
2.2.2 流动相的选择流动相不仅会直接影响目标物的峰形,还会对目标物的保留行为产生影响,此外流动相也是影响质谱灵敏度的因素之一。在选定的色谱柱上,以乙腈为有机相,比较了盐类缓冲体系(5 mmol/L 乙酸铵溶液、5 mmol/L 甲酸铵溶液)、5 mmol/L 乙酸铵溶液(含0.2%甲酸溶液)、5 mmol/L甲酸铵溶液(含0.2%甲酸溶液)、0.2%甲酸溶液和0.2%乙酸溶液6种水相对8种替丁类药物的分离情况。结果显示,雷尼替丁、法莫替丁及硫替丁的峰形可能是受铵盐影响,在盐类缓冲体系下存在色谱峰过宽或分叉等情况;盐类缓冲体系中添加0.2%甲酸后,法莫替丁及硫替丁的峰形得到改善,但雷尼替丁的峰形无明显改善,原因可能是替丁类化合物结构中N 原子上的孤电子对能接受质子,对流动相pH 值较敏感,其中以雷尼替丁最为明显;0.2%甲酸溶液和0.2%乙酸溶液两种水相体系下,雷尼替丁的峰形均得到改善,相对而言,0.2%甲酸溶液体系下雷尼替丁的峰形更尖更窄,推测是因为甲酸的酸性比乙酸强所致。经反复实验,以0.2%甲酸溶液为水相,乙腈为有机相,采用梯度洗脱,8 种替丁类药物可获得较佳分离效果且保留时间分布均匀,色谱峰无拖尾,质谱灵敏度良好。综合考虑,最终确定的色谱分离条件如“1.4.1”所示。在该条件下,8种替丁类药物的色谱峰形良好,色谱保留适中且灵敏度高(见图2)。

图2 8种替丁类药物混合标准溶液的提取离子色谱图Fig.2 MRM chromatograms of mixed standard solution of eight tidine drugs
2.3 样品前处理条件的优化
本实验前处理条件的优化主要在于提取溶剂的选择。待测化合物极性和样品基质类型决定了提取溶剂的选择[11]。实验涉及的8种替丁类药物结构和性质相近,易溶于甲醇和乙腈等;化妆品基质复杂,不同基质类型的化妆品适用的提取溶剂也不尽相同,通用型提取溶剂主要有甲醇和乙腈等。以乙腈作为提取溶剂时,膏霜类等复杂基质样品不易分散,而用少量水分散后再加入乙腈则具有更好的润湿性,增加了提取溶剂的渗透性,同时也使基质分散更均匀,对大部分化合物的提取效果更好[12]。此外,超声波辅助提取广泛用于化妆品的样品前处理,水剂类、膏霜类、乳液类、凝胶类和粉剂类等绝大多数化妆品样品均可用该法进行前处理[13-14]。在此基础上,以5 种不同类型化妆品(包括水剂类、膏霜类、乳液类、凝胶类和粉剂类)为样品基质,分别比较了甲醇和水-乙腈(2∶8,体积比)作为溶剂,超声波辅助提取时,各替丁类化合物的回收率(见图3)。结果显示,采用两种提取溶剂超声提取15 min 条件下,8 种替丁类药物的回收率相差不大,回收率均较高。考虑到乙腈沉淀大分子杂质(如硅油、蜡质等)、增溶剂(如色素及蓖麻油等)等的效果优于甲醇,基于对仪器、色谱柱等的保护,选用水-乙腈(2∶8)作为提取溶剂。
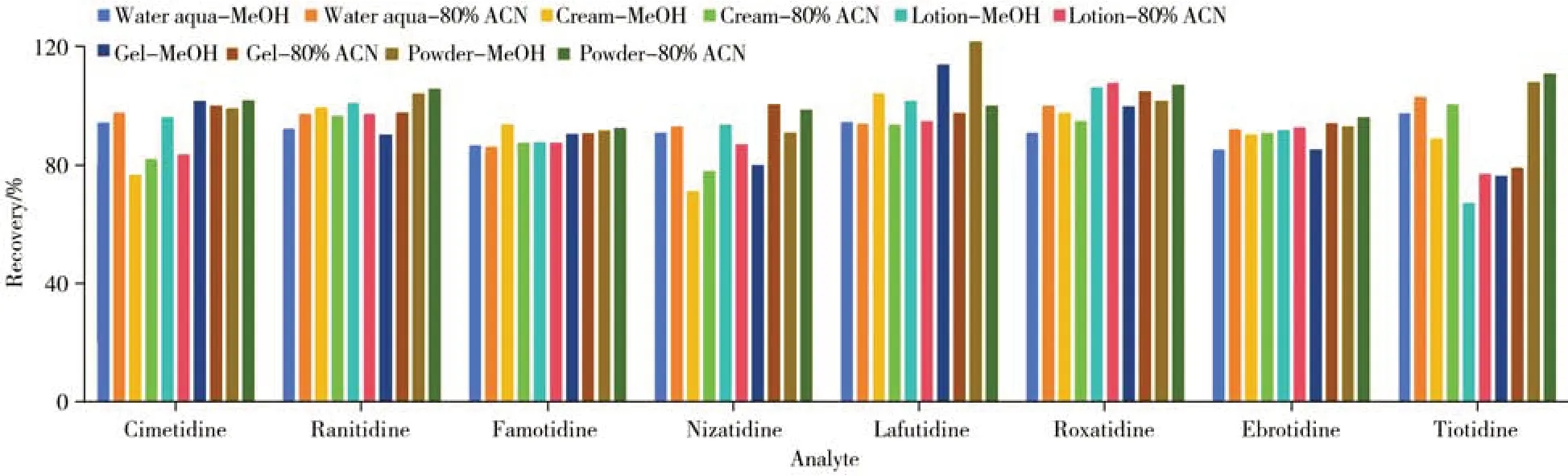
图3 提取溶剂对8种替丁类药物回收率的影响Fig.3 Effect of extraction solvents on recoveries of eight tidine drugs
2.4 基质效应评价
基质效应是液相色谱-质谱分析过程中不可忽略的关键因素,不仅直接影响方法的灵敏度,还影响方法的准确度和精密度。基质效应通常采用公式ME=B/A进行计算,其中B为空白基质溶液作为溶剂配制的标准曲线的斜率,A为纯溶剂配制的标准曲线的斜率[15];ME值越接近100%,表明基质效应越可忽略不计。5种不同类型化妆品基质(包括水剂类、膏霜类、乳液类、凝胶类和粉剂类)中8种替丁类药物的基质效应见表2。结果表明,乙溴替丁在大多数基质中存在一定的基质效应,硫替丁在水剂类基质中也存在一定的基质效应。减少基质效应的方法通常包括增加净化步骤、稀释样品溶液、配制空白基质匹配标准溶液、采用同位素内标物校正以及优化色谱-质谱条件等[16]。为保证定量结果的准确性,综合考虑成本、操作方便性,本文选择基质匹配标准溶液法降低基质效应的影响。
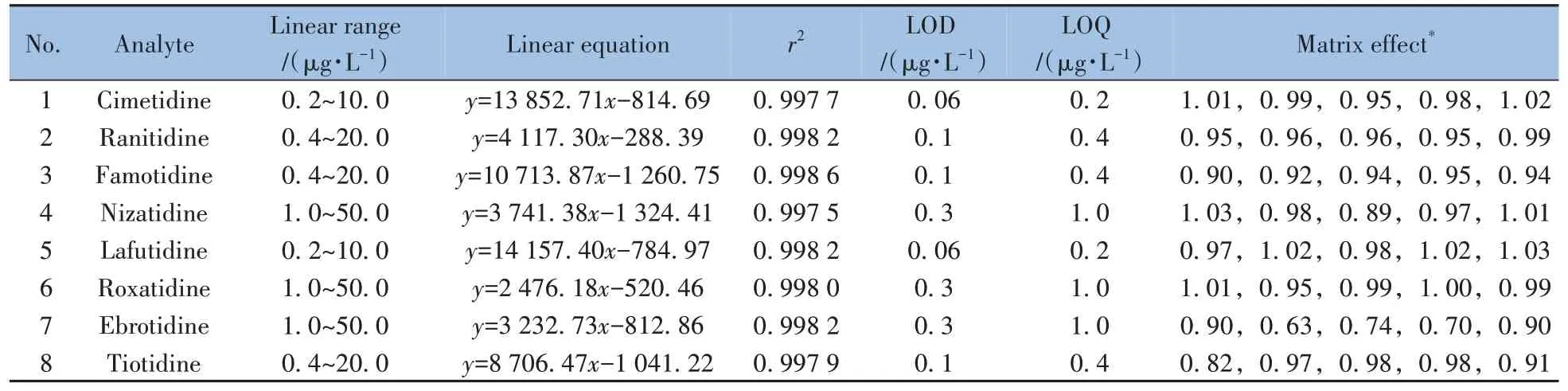
表2 8种替丁类药物的线性关系、检出限、定量下限和基质效应Table 2 Linear relationship,LODs,LOQs and matrix effects of eight tidine drugs
2.5 线性范围、检出限与定量下限
用空白基质提取液将8 种替丁类药物稀释成系列混合标准溶液,按“1.4”的仪器条件进行测定,以各替丁类药物的质量浓度为横坐标(x,μg/L),峰面积为纵坐标(y),进行线性回归分析。8种替丁类药物均获得良好的线性关系,相关系数(r2)为0.997 5~0.998 6。采用标准添加法确定各替丁类药物的检出限(S/N≥3)和定量下限(S/N≥10)分别为0.06~0.3 μg/L和0.2~1.0 μg/L(见表2)。
2.6 回收率与相对标准偏差
准确称取空白水剂类、膏霜类、乳液类、凝胶类和粉剂类基质化妆品各0.2 g,进行3 个加标水平的回收实验,每个水平按“1.3”和“1.4”方法平行测定6 次,计算各替丁类药物的平均回收率和相对标准偏差(RSD,n=6)。由表3可见,水剂类、膏霜类、乳液类、凝胶类和粉剂类基质的平均回收率分别为80.7%~106%、90.1%~102%、82.6%~101%、82.0%~110%和86.6%~101%,均在80%~115%之间;RSD 分别为0.70%~7.0%、1.0%~5.7%、2.1%~6.9%、0.90%~7.4%和1.7%~ 7.9%,均小于10%。结果表明,本方法的准确度和精密度良好,适用于5种基质化妆品中8种替丁类药物的检测。

表3 8种替丁类药物在不同基质中的平均回收率和相对标准偏差(n=6)Table 3 Average recoveries and relative standard deviations(RSDs) of eight tidine drugs in different matrixes(n=6)
2.7 稳定性
分别在空白水剂类、膏霜类、乳液类、凝胶类和粉剂类基质化妆品中添加低、中、高3个水平的8种替丁类药物,同一日内连续6 次(0、5、10、15、20、24 h)测定,以峰面积计算8 种替丁类药物在5种基质中的RSD。结果表明,5 种不同空白基质中,8 种替丁类药物在3 个添加水平下的RSD 分别为1.2%~9.0%、0.80%~7.3%和1.1%~6.7%,本方法具有较好的稳定性。
2.8 实际样品测定
采用本文所建方法对客户委托送检的100批次化妆品进行分析,在某款祛痘凝胶中检出西咪替丁,含量为28.2 μg/g。该阳性样品的总离子流图及西咪替丁的MRM色谱图见图4。

图4 阳性样品的总离子流图(A)及西咪替丁的MRM色谱图(B、C)Fig.4 TIC chromatogram(A) and MRM chromatograms(B,C) of cimetidine in a positive sample
3 结 论
本研究建立了同时快速测定5种不同类型化妆品基质(包括水剂类、膏霜类、乳液类、凝胶类和粉剂类)中8 种替丁类药物的UHPLC-MS/MS 法。方法前处理简单方便、灵敏度高、可行性强,可用于多种基质类型化妆品中替丁类药物的检测。该方法可作为政府部门有效的技术支持和补充方案,为监测化妆品中非法添加药物提供科学依据,便于化妆品市场的监管。
猜你喜欢
中国典型病例大全(2022年7期)2022-04-22 00:50:34
广东药科大学学报(2020年2期)2020-03-03 15:57:01
中国蜂业(2018年4期)2018-05-09 06:25:08
当代化工研究(2016年6期)2016-03-20 16:21:46
中国卫生标准管理(2015年13期)2016-01-15 02:58:25
中国卫生标准管理(2015年25期)2016-01-14 09:29:24
湖南中医药大学学报(2015年1期)2016-01-06 01:06:36
中国卫生标准管理(2015年15期)2015-01-26 20:32:38
无机化学学报(2014年3期)2014-02-28 17:30:58
河南科技(2014年12期)2014-02-27 14:10:32
