
过渡金属元素掺杂对SmCo3 合金结构和磁性能影响的第一性原理计算*
2024-02-21 13:12:30严志方诚王芳许小红
物理学报 2024年3期
严志 方诚 王芳 许小红
(山西师范大学化学与材料科学学院,磁性分子与磁信息材料教育部重点实验室,材料科学研究院,太原 030031)
1 引言
Sm-Co 基稀土永磁合金具有优异的高温磁性能,被广泛应用于航空航天、新能源和国防军工等领域,尤其是在工作温度超过500 ℃的条件下有着无可替代的地位[1–6].因此,其从1967 年[7]发现至今得到了广泛的关注和研究.Sm-Co 基稀土永磁合金具有多种构型,如SmCo3,SmCo5和Sm2Co17等.SmCo3晶体结构可以由SmCo5晶体结构通过Sm 原子替代一些Co 原子位点得到[8],其轴比c/a>4.8[9],具有较大的晶格各向异性,有利于磁性能的提高.但是由于其较低的饱和磁化强度[10]限制了最大磁能积,从而阻碍了实际应用.
前人的研究表明,掺杂合金元素是增强Sm-Co基稀土永磁合金的稳定性和磁性能的理想途径之一[11–14].实验方面,Guo 等[15]采用Mn 掺杂SmCo7纳米晶结构,发现Mn 元素掺杂既可以提高结构稳定性,又可以提升体系综合磁性能,并探究出最佳掺杂浓度.Wang 等[16]以Fe,Ni 和Nb 为掺杂元素,发现元素Fe 和Ni 可以有效提高Sm5Co19基纳米合金的综合磁性能.理论计算方面,Das 等[17]基于第一性原理计算,在SmCo5体系中用Fe 替代Co,对不同浓度Fe 掺杂SmCo5合金的晶体结构稳定性、磁化强度和磁晶各向异性能进行研究,计算发现高浓度Fe 的掺杂不利于晶体结构稳定性,同时使磁晶各向异性能下降,但是可以提高体系的饱和磁化强度,Liu 和Altounian[18]也得出类似的结论.Landa 等[19]通过第一性原理计算,在SmCo5体系中用Fe 替代大部分Co 并使用Ni 作为热力学稳定剂,产生了具有优异磁性的SmCoNiFe3合金,磁化强度与SmCo5合金相比从8.27 μB增至10 μB,且具有高的居里温度和强的磁各向异性,其磁各向异性能大小约为Nd2Fe14B 合金的两倍.无独有偶,Antoniou 等[20]也同样发现在SmCo5体系中掺杂Ni 可以提高掺杂体系的结构稳定性.综上所述,实验探索和理论计算两方面均表明掺杂过渡元素对于提升Sm-Co 基稀土永磁合金的结构稳定性和磁性能有着非常重要的作用.而第一性原理计算可以从体系的能量、晶体结构和电子结构等微观层面分析材料的稳定性和相关磁性能,从而筛选出有利于提升Sm-Co 基稀土永磁合金综合磁性能的掺杂元素,有望从理论上为解决国内外稀土永磁新材料与新产品研发周期长和成本高的缺点提供有益指导.
目前,国内外对于SmCo3体系的实验探索和理论研究还非常少,尤其是掺杂过渡元素的理论研究,且掺杂元素种类和掺杂浓度在SmCo3合金中的优先占位情况和对磁性能的影响机制尚不明确.本文基于第一性原理计算,在SmCo3体系中设计多种过渡元素的掺杂构型.通过对掺杂体系的晶格常数、掺杂元素的优先占位、电子结构和磁矩等方面的计算,得出了过渡元素掺杂对SmCo3体系结构稳定性和磁性能的作用关系,并揭示了其微观机理.此外,基于掺杂结构稳定性、磁性能和掺杂元素的浓度三方面作用因素,筛选出了最佳的掺杂元素和其掺杂浓度,为新型高性能Sm-Co 基永磁合金的研究和开发提供了有益的理论指导.
2 模型构建和计算方法
2.1 模型构建
SmCo3的晶体结构属于PuNi3型三方(菱方)晶系,空间群为R¯3m.如图1(a)所示,其单胞中有36 个原子,3 个Sm 原子占据3a晶格位置,6 个Sm 原子占据6c晶格位置,3 个Co 原子占据3b晶格位置,6 个Co 原子占据6c晶格位置,18个Co 原子占据18h晶格位置.在三维坐标系中,5 个不等价的晶格位置坐标分别为: Sm(3a)原子坐标为(0,0,0);Sm(6c)原子坐标为(2/3,1/3,0.193699);Co(3b)原子坐标为(1/3,2/3,1/6);Co(6c)原子坐标为(1/3,2/3,0.332163);Co(18h)原子坐标为(2/3,5/6,0.253176),其实验晶格常数为a=b=5.050 Å,c=24.590 Å,c/a=4.8693[10].掺杂过渡元素以Fe,Ni,Cu 和Zr 为代表,建立未掺杂的SmCo3晶胞(1×1×1)和掺杂原子百分比为3.7%的Sm9Co26M(M=Fe,Ni,Cu,Zr)的模型,而且在计算过程中考虑掺杂元素占据Co 的3b,6c和18h的晶格位置3 种情况,如图1 所示.
2.2 计算方法及相关参数
本研究的计算都是在VASP (Viennaab-initiosimulation package)[21,22]中的密度泛函理论(DFT)框架下,通过平面波基投影增强波(PAW)方法[23,24]进行的.使用广义梯度近似下的平面波(PBE)泛函[25,26].平面波截断能设置为500 eV,原子的自洽总能量收敛标准和几何(弛豫)结构力的收敛标准分别设为10–5eV 和0.01 eV/Å,K点设置为9×9×3.在结构弛豫过程中,原子位置以及晶胞的形状和体积都被允许弛豫.为了更好地描述d 轨道和f 轨道电子,本文采用Dudarev 提出的DFT+U方法[27],对Sm,Fe,Co,Ni,Cu 和Zr 原子的有效U值分别设置为4.70 eV,1.83 eV,2.22 eV,1.61 eV,4.80 eV 和1.50 eV,这些U值的选取文献[28,29].计算时各原子的电子组态分别为: Sm-5s25p65d16s2,Fe-3d64s2,Co-3d74s2,Ni-3d84s2,Cu-3d104s1和Zr-4d25s2.
Sm 元素的赝势选择Sm_3 进行计算,也是VASP 官方推荐使用的赝势[30,31].该赝势将4f 电子作为原子核内电子,不作为价电子,本文研究的是掺杂过渡元素种类和浓度对SmCo3体系结构稳定性和磁矩的影响,而不需要考虑4f-3d 轨道电子相互作用的影响,因此选择该赝势是合理的,也是研究Sm-Co 合金体系常用的处理方式.
3 计算结果分析与讨论
3.1 掺杂元素占位与结构稳定性
首先,对掺杂前后的模型进行结构优化,SmCo3晶胞的晶格常数a=b=5.0123 Å,c=24.6424 Å,体积V=536.17 Å3,与实验晶格参数a=b=5.050 Å,c=24.590 Å,V=543.09 Å3[10]相吻合.如表1 所列,优化后的晶格常数与实验结果相差仅1%左右,计算得到的SmCo3晶胞体积比实验体积约小2%,说明计算结果是可靠的.由于掺杂元素的原子半径不同,SmCo3掺杂体系的c/a略有不同,且掺杂体系的晶胞体积随着掺杂元素的原子半径的增大而增大.
为研究掺杂元素种类和占位对晶体结构稳定性的影响,计算了掺杂Sm9Co26M体系的替代能,具体公式为[32]
式中,E(Sm9Co27–nMn)表示掺杂体系的总能量,E(SmCo3)表示未掺杂体系的总能量,E(Co)和E(M)分别表示Co 和过渡金属元素M处于单质状态时每个原子的基态能量,n表示原子个数.替代能为负值表示该元素掺杂较容易,且有利于提升掺杂体系的结构稳定性;反之正值则表示该元素掺杂较难同时不利于体系结构稳定性.元素在不同晶格位置取代Co 原子时,优先占据替代能较低的位点,不同掺杂体系的替代能如图2 所示.从图2 可以发现Fe,Ni,Cu 和Zr 都优先占据18h位点.替代能越低,晶体结构越稳定,Ni,Cu 和Fe 的掺杂提高了SmCo3结构的稳定性,与之前的研究结果一致[17,20];而Zr 的掺杂则不利于SmCo3结构的稳定性.插图显示掺杂元素在优先占位的体系替代能,由此可以得出掺杂元素对SmCo3结构的稳定能力由大到小依次为: Ni,Cu,Fe,Zr.
3.2 差分电荷密度分析
为阐明SmCo3掺杂M(M=Fe,Ni,Cu,Zr)后体系中各类原子间的电荷转移机制,计算了不同M原子掺杂在18h晶位(001)晶面的差分电荷密度,结果如图3 所示.图3(c),(d)中Ni 和Cu 原子替代Co 原子后,在Co-Ni 和Co-Cu 原子间聚集大量电子,且倾向于向掺杂原子Ni 和Cu 原子转移.这表明图3(c),(d)中Co-Ni 和Co-Cu 原子间金属键得到明显加强,且大于图3(a)中本身的Co—Co 键,从而提升了掺杂体系稳定性;图3(e)中Zr原子替代Co 原子后,Zr 原子周围表现为明显的电子缺失,且在Co-Zr 原子间的差分电荷密度没有重叠,表明Co-Zr 原子间的相互作用减弱,从而降低了掺杂体系稳定性;图3(b)中Fe 原子替代Co 原子与未掺杂图3(a)中Co 原子之间电荷密度差异并不明显;上述分析与3.1 节中替代能计算结果定性一致.造成原子间电子转移情况的不同主要归因于每个原子的电负性各不相同,从大到小排列顺序依次为Ni,Cu,Co,Fe,Zr.
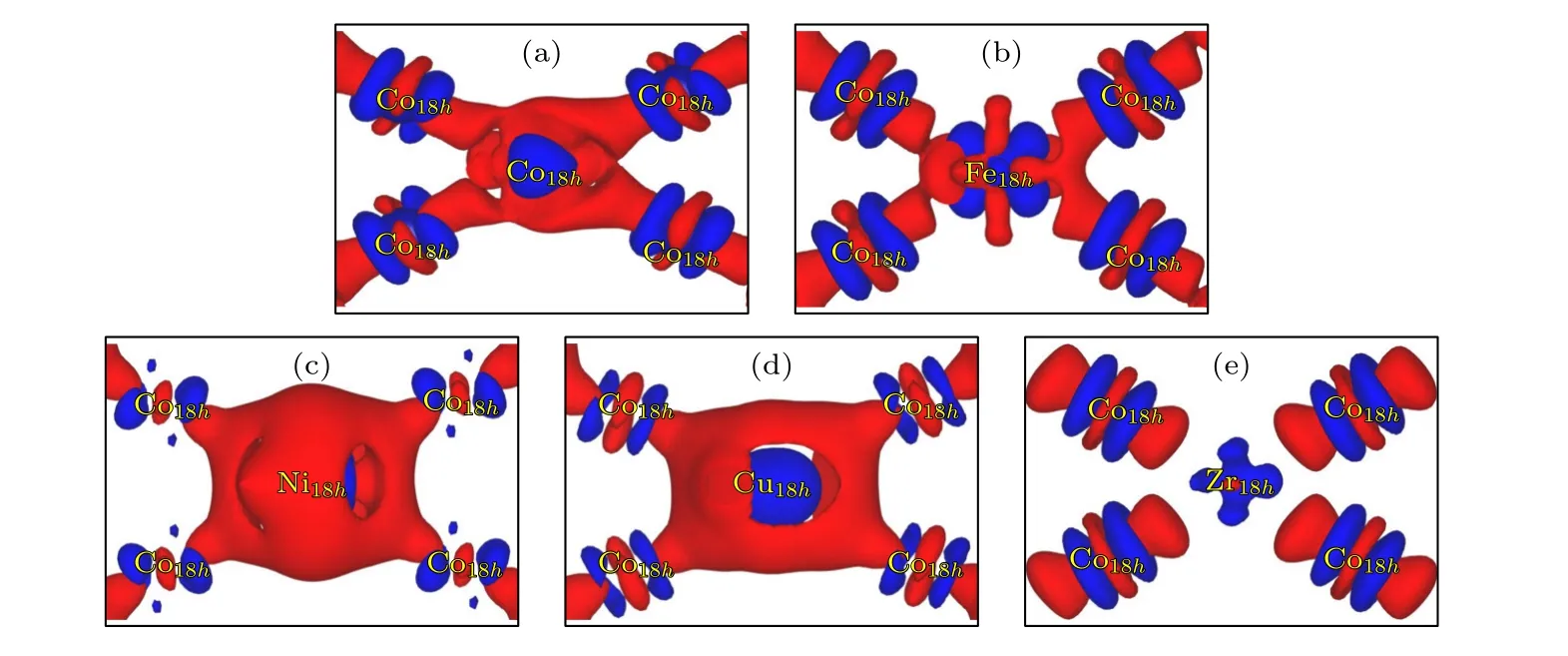
图3 (001)面上(a) SmCo3 和(b)—(e) Sm9Co26M (M=Fe,Ni,Cu,Zr)的差分电荷密度;红色表示电荷的积累,蓝色表示电荷的损耗Fig.3.Difference charge density of (a) SmCo3 and (b)—(e) Sm9Co26M (M=Fe,Ni,Cu,Zr) on (001) plane;the red indicates enrichment of electrons,blue indicates loss of electrons.
3.3 掺杂元素种类对基体总磁矩的影响
图4 展现了各个掺杂体系总磁矩的计算结果,图中虚线表示未掺杂SmCo3体系的总磁矩为38.74 μB,与实验值37.35 μB接近[10].导致理论和实验磁矩的微小差别可能有以下两种原因: 一是在实验制备SmCo3合金的过程中,并不能完美地控制Sm:Co 的含量比为1∶3;另一种是计算中只考虑了原子的自旋磁矩,而忽略了原子的轨道磁矩.从图4 可知: Ni,Cu 和Zr 元素的掺杂使体系的总磁矩减弱,主要原因是Cu 和Zr 元素是非磁性元素;而Ni 作为磁性元素并没有增大体系的总磁矩,这与传统磁性元素[12,13]掺杂提升体系饱和磁化强度的认识不符;磁性元素Fe 掺杂使体系的总磁矩大幅增大.此外,当M原子在不同晶格位置取代Co原子时,对总磁矩的影响也各不相同.从图4 可以看出,掺杂元素Fe,Ni,Cu 在不同位置总磁矩的差异很小.但是掺杂元素Zr 的体系在取代不同位置的Co 时,对总磁矩的影响很大,其中掺杂在3b,6c和18h晶格位置体系的总磁矩分别为38.73 μB,37.43 μB和37.02 μB.主要原因是Zr 的原子半径远远大于Co 原子和不同位置的晶格间隙不同,Zr原子掺杂后造成晶格内附近的Co 原子位置发生不同程度的位移,其中3b晶格位置间隙最小,使周围Co 原子之间距离减小,增强了其周围Co 原子的磁矩,因此对体系总磁矩的减小程度产生了一定的补偿作用.
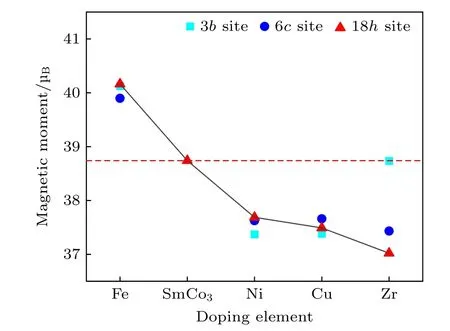
图4 SmCo3 和Sm9Co26M (M=Fe,Ni,Cu,Zr)的总磁矩;红色虚线表示未掺杂SmCo3 体系的总磁矩为38.74 μBFig.4.Total magnetic moments of SmCo3 and Sm9Co26M(M=Fe,Ni,Cu,Zr);the red dotted line indicates that the total magnetic moment of the undoped SmCo3 system is 38.74 μB.
3.4 偏态密度分析
为进一步揭示掺杂元素对SmCo3体系总磁矩影响的微观机制,选择最稳定的掺杂结构(18h)进行元素电子的偏态密度(partial density of state)分析,如图5 所示.灰色阴影代表上下自旋态密度对称部分,而红色阴影代表不同部分.体系的净磁矩源于上下自旋态密度的不对称性.如图5(a),(b)所示,Fe 在18h晶格位置的上下自旋态密度对称性远低于Co,说明Fe 原子磁矩大于Co 原子磁矩,所以Fe 掺杂可以提升体系的总磁矩;图5(c)表示Ni 元素在18h晶格位置的偏态密度,由于其费米能级附近上下自旋电子数较少,且上下自旋态密度对称性较高,说明其磁矩很小,远远小于Co 元素在18h晶格位置的磁矩,因此Ni 的掺杂降低了体系的总磁矩;图5(d),(e)分别表示Cu 元素和Zr元素在18h晶格位置的偏态密度,两者上下自旋态密度都高度对称.前者在费米能级附近上下自旋电子数稀少,对应红色区域表示的自旋差值部分也非常少,且在较深能级处出现了上下自旋对称灰色部分,说明Cu 原子本身具有的磁矩非常小.而后者在费米能级附近上下自旋电子数相对较多,且红色自旋差值部分也较大,但是远远小于Co 元素在该位置的红色区域,说明Zr 原子具有一定大小的磁矩但小于Co 原子的磁矩.原子Cu,Zr 与原子Co的本征自旋磁矩方向相反,造成体系总磁矩下降,但是Zr 原子磁矩大于Cu 原子磁矩,所以Zr 原子掺杂对体系的总磁矩影响更大.经过上述PDOS分析,与上文中磁矩计算结果一致.
3.5 元素的筛选
如图6 所示,给出不同掺杂体系最稳定掺杂结构(18h)的替代能和总磁矩.蓝色虚线代表未掺杂SmCo3体系的总磁矩,红色虚线代表未掺杂SmCo5体系的替代能,其值为0.从图6 可以看出,红线左侧掺杂元素的体系具有较低的替代能,有利于PuNi3型晶体结构稳定性.掺杂体系替代能从小到大排列依次是Ni,Cu,Fe 以及不利于结构稳定的Zr.所以从结构稳定性方面选择的最佳元素是Ni,Cu 次之,最后是Fe.另一方面,蓝色虚线以上的掺杂体系,只有Sm9Co26Fe 体系总磁矩比未掺杂的总磁矩高,而蓝色虚线以下的体系总磁矩则比未掺杂的低.因此,在4 种过渡金属元素中只有Fe 元素掺杂既有利于提升结构稳定性,又能提高SmCo3体系的总磁矩.
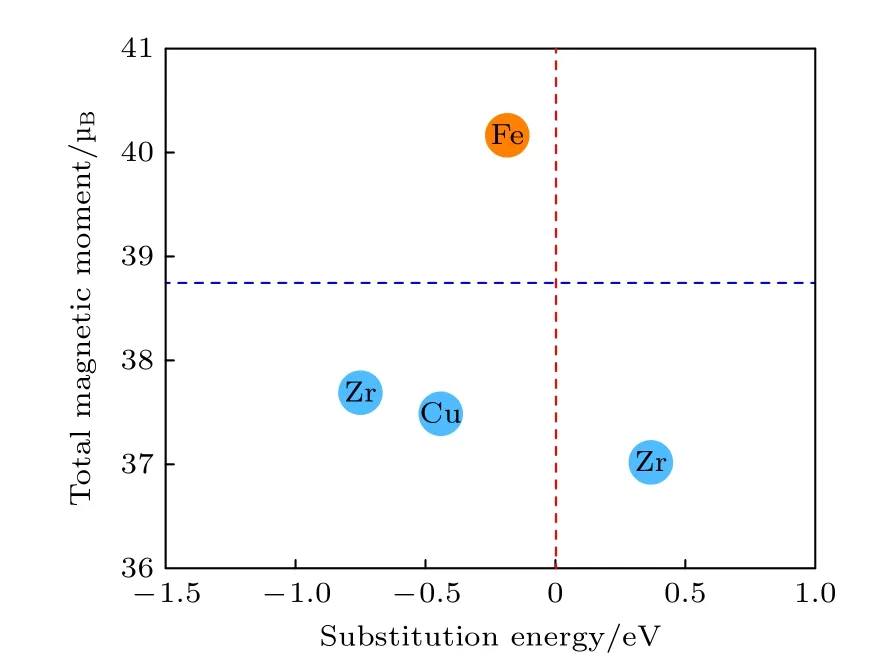
图6 Sm9Co26M (M=Fe,Ni,Cu,Zr)合金的替代能和总磁矩Fig.6.Substitution energy and total magnetic moment of the Sm9Co26M (M=Fe,Ni,Cu,Zr) alloys.
3.6 掺杂元素浓度对结构稳定性和总磁矩的影响
通过上述对4 种过渡元素掺杂SmCo3体系进行系统计算分析,得到掺杂不同种类过渡元素对SmCo3体系结构稳定性、总磁矩和电子结构可以产生很大的影响.在实际的Sm-Co 稀土材料研发中,仅仅了解掺杂元素的种类并不足以确保材料性能的最优化.为实现最佳效果,必须精确调控各组成的含量.如在SmCo5体系中掺杂Fe 时,在合适的浓度范围内可以提高饱和磁化强度和居里温度,但过量掺杂会导致饱和磁化强度、居里温度和结构稳定性下降,使其综合磁性能受到影响[17,18,33].为了进一步分析4 种过渡元素掺杂浓度对体系的影响,并预测合适的掺杂浓度范围.本文参照实验可行性掺杂浓度范围[34],在SmCo3单原胞中,掺杂原子百分比为0—22.22%范围内,根据原子个数配比设置了计算原子百分比分别为x=3.7%,7.4%,11.11%,14.81%,18.52%,22.22%的掺杂体系.
如图7 所示,分别计算了SmCo3–xMx体系中替代能和体系总磁矩随着掺杂浓度的变化趋势(结构都是最稳定的),红色(黑色)折线表示体系总磁矩(替代能)随着掺杂浓度的演化关系.如图7(a)所示,在SmCo3–xFex体系中,可看出随着Fe 掺杂浓度的增大,掺杂体系的总磁矩也随之增大,同时替代能随之降低,表明掺杂体系结构也越来越稳定;图7(b),(c)分别表示SmCo3–xNix和SmCo3–xCux体系的演化情况,可以看出,随着掺杂浓度的增大,体系的替代能几乎呈线性方式下降,表明体系的结构越来越稳定,但总磁矩下降;图7(d)表示SmCo3–xZrx体系,伴随着掺杂浓度的增大,体系的替代能几乎呈线性方式上升,表明体系的结构越来越不稳定,同时掺杂体系的总磁矩也在下降.综合4 种SmCo3基掺杂体系随着掺杂浓度变化的替代能和总磁矩特点,可以预测在掺杂原子百分比为0—22.22%范围内,Fe 元素为最佳的掺杂原子,与3.5 节叙述结果相对应,且其最优的掺杂原子百分比为18.52%左右(考虑到实验的可行性掺杂浓度).

图7 SmCo3–xMx (M=Fe (a),Ni (b),Cu (c),Zr (d))体系替代能和体系总磁矩随掺杂浓度的变化Fig.7.Relationship between the substitution energy and total magnetic moment of SmCo3–xMx (M=Fe (a),Ni (b),Cu (c),Zr(d) system with doping concentration.
4 结论
采用第一性原理计算方法,研究过渡金属M=Fe,Ni,Cu,Zr 掺杂的SmCo3型永磁合金的结构稳定性和磁性能.主要结论如下:
1)通过计算各掺杂体系的替代能可得,过渡元素Fe,Ni,Cu 和Zr 都优先占据18h晶格位置,元素Ni,Cu 和Fe 的掺杂可以降低掺杂体系的替代能,从而提升SmCo3体系结构稳定性;而元素Zr 掺杂不利于体系结构稳定性.
2)添加磁性元素Fe 可以使体系总磁矩从38.74 μB增至40.16 μB,而添加磁性元素Ni 却使体系总磁矩从38.74 μB降低到37.67 μB.这说明在SmCo3体系并不是所有的铁磁元素都可以增加体系的总磁矩.掺杂Cu 和Zr 这种非磁性元素通常会在一定程度上减弱SmCo3体系的总磁矩.
3)在SmCo3单原胞中,掺杂原子百分比为0—22.22%范围内,通过计算获得了SmCo3不同掺杂体系结构稳定性和磁矩随着掺杂元素浓度变化规律.最后筛选出既能提高掺杂体系结构稳定性,又可以提升磁性能的元素: Fe,并预测了其最合适的掺杂原子百分比为18.52%.
猜你喜欢
数学物理学报(2019年5期)2019-11-29 07:46:50
数学物理学报(2017年5期)2017-11-23 07:51:09
军事文摘·科学少年(2017年4期)2017-06-20 23:22:10
材料科学与工程学报(2016年2期)2017-01-15 13:34:42
潍坊学院学报(2016年6期)2016-04-18 13:56:55
深圳大学学报(理工版)(2015年6期)2015-11-26 12:33:48
警察技术(2015年4期)2015-02-27 15:37:51
长江大学学报(自科版)(2014年1期)2014-03-20 13:20:12
河南科技(2014年23期)2014-02-27 14:18:52
物理通报(2011年9期)2011-01-24 07:39:36
