
血浆置换治疗儿童 AQP4-IgG阳性视神经脊髓炎1例及文献分析*
2024-02-16 14:24:52张耀元李树军麻赫锴任瑞娟王志远
医学理论与实践 2024年1期
张耀元 李树军 麻赫锴 任瑞娟 王志远
新乡医学院第一附属医院儿科,河南省卫辉市 453100
视神经脊髓炎谱系疾病(Neuromyelitis opticaspectrum disorders,NMOSD)是一种主要由体液免疫参与的抗原—抗体介导中枢神经系统炎性脱髓鞘疾病谱,多见于青壮年患者,女性居多,儿童病例占3%~5%[1]。国际视神经脊髓炎诊断小组(International Panel for Neuromyelitis Optica Diagnosis,IPND)2015年提出广义NMOSD概念,包括视神经炎、急性脊髓炎、极后区综合征、间脑综合征、急性脑干综合征、急性大脑综合征在内6项核心临床特征(Core clinical characteristics ,CCC),并以水通道蛋白4抗体(AQP4-IgG)作为分层,制定AQP4-IgG阳性、AQP4-IgG阴性或AQP4-IgG未知状态诊断标准[2]。该病具有高致残、高复发、多样化的特点,其中极后区综合征多以顽固性呃逆、恶心、呕吐等消化道症状起病,临床中容易误诊。本文报道1例14岁女性患者,以消化系统疾病起病,最终确诊为以极后区综合征为首发表现NMOSD,应用激素、人免疫球蛋白治疗效果差,后给予血浆置换等综合治疗后达到临床缓解,报道如下。
1 病例资料
患儿女,14岁,以“恶心、呕吐4d”为主诉入院。4d前出现恶心、呕吐,伴食欲、进食差,腹部不适感,伴阵发性皮肤瘙痒感,发作顺序为颈部—躯干—双上肢,伴双上肢不自主颤动。入住消化科。自发病以来,体重较前下降约1kg(2周内)。既往史、个人史、家族史无殊。给予对症支持治疗3d,恶心、呕吐缓解,出现反复头痛、颈部疼痛、皮肤瘙痒感、肢体抖动,头颅MRI:未见明显异常。颈椎+胸椎MRI示:延髓下缘—颈段脊髓内见结节状及条片状等T1长/稍长T2信号。胸1~8椎体段脊髓内见细条状等T1长/稍长T2信号(见图1),考虑存在脊髓炎。患儿出现一过性意识丧失、面色苍白,心电示室性心动过速,急转入PICU治疗,患儿意识恢复,心律转为窦性。体格检查:T 36.3℃,P 75次/min,R 20次/min,W 51kg,BP 117/91mmHg(1mmHg=0.133kPa),神志清楚,精神差,颈部活动受限,双侧瞳孔等大等圆,对光反射灵敏。颈胸段椎体轻压痛,心、肺、腹部查体未见明显异常;神经系统查体:角膜反射存在,腹壁反射消失,约平寰椎至第1肋间区域感觉障碍:痛温觉降低,自感皮肤瘙痒,余部位痛温觉正常。位置觉正常,浅感觉(痛温觉)存在,肌张力正常,左上肢肌力4级,余肢体肌力5级。生理反射:膝腱反射、跟腱反射活跃;病理反射:双侧Babinski征阳性。脑膜刺激征:颈强直(+),Kernig(-)。Glasgow评分:睁眼3分(语言刺激时)、最佳运动反应5分(因局部疼痛而动)、最佳语言反应5分(能定向说话)。临床扩展致残量表评分(Expanded Disa-bility Status Scale,EDSS)8.0分;辅助检查:血常规、电解质、肝肾功、血糖、乳酸、血氨、凝血功能、动脉血气分析,免疫五项在正常范围;抗髓过氧化物抗体、抗蛋白酶3抗体、中性粒细胞胞浆抗体均阴性;抗甲状腺抗体阳性;抗核抗体(IIF)阳性(核颗粒型),抗Ro52kd抗体(LIA)阳性,抗ds-DNA(IIF),抗核小体抗体(LIA),抗ds-DNA(LIA),抗组蛋白抗体(LIA),抗SmD1抗体(LIA),抗U1-snRNP抗体(LIA),抗SSB/R60抗体(LIA),抗SSB/La抗体(LIA),抗Scl/70抗体(LIA),抗着丝点抗体(LIA),抗J01抗体(LIA)均阴性。ANA滴度检测:ANA滴度1∶80阳性,ANA滴度1∶160弱阳性。淋巴细胞亚群测定在正常范围。脑脊液:压力170mmH2O(1mmH2O=9.8Pa);常规:无色、清亮、无凝集、潘氏试验弱阳性(±),有核细胞计数100×106/L分类:多叶核20%,单个核80%;脑脊液生化:Cl-119.0mmol/L,GLU 3.33mmol/L,ADA 2.00U/L,PRO 396.2mg/L。脑脊液免疫球蛋白35.6mg/L。脑脊液:PCR-HSV·Ⅰ阴性,墨汁染色未找到隐球菌。脑脊液培养无阳性结果。血清抗AQP4抗体IgG:1∶32,抗MOG抗体IgG阴性,抗GFAP抗体IgG阴性。

图1 颈椎+胸椎3.0磁共振成像(MRI)
临床诊断:视神经脊髓炎谱系疾病(NMOSD)。给予甲泼尼龙(0.5g,q12h×5d)冲击,患儿症状未见缓解,继续应用人免疫球蛋白(20g,qd×5d)治疗,头颈部疼痛和肢体异常有加重趋势,复查磁共振成像(MRI)平扫+增强:延髓—颈—胸3椎体水平脊髓增粗,髓内可见条片状、不均匀稍长T2信号影(见图2),复查脑脊液:压力140mmH2O;潘氏试验阴性,有核细胞计数20×106/L,多叶核细胞0%,单个核细胞0%。脑脊液生化正常。继续甲泼尼龙治疗6d(0.5g,qd×3d,0.25g,qd×3d);患儿颈胸段疼痛持续存在,伴间断头痛、皮肤瘙痒感,间断存在四肢不自主抖动,夜间噩梦增多,出现幻视。脑电图检未见异常;眼部检查:双眼眼底视盘色正常,边界清,无水肿,视网膜血管走形及比例均正常,黄斑区未见异常。视神经核磁未见异常。视力:双眼0.8。视觉诱发电位:左侧VEP-P100潜伏时延长,右侧VEP-P100潜伏时未见异常。
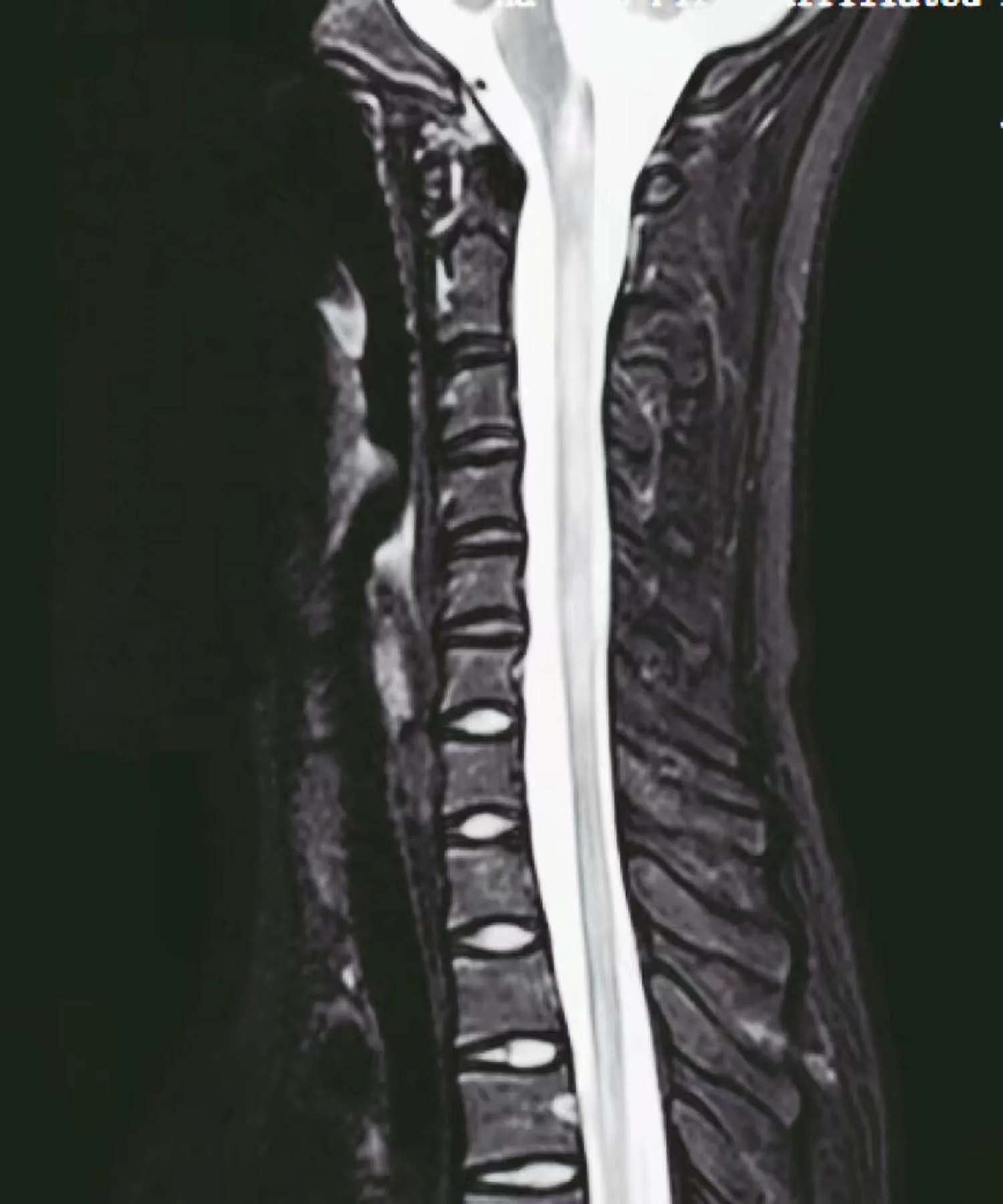
图2 头颈胸腰骶3.0磁共振成像(MRI)平扫+增强
患儿临床症状以及影像学提示有加重趋势,EDSS评分较前增加至8.5分,免疫治疗效果差,进行血浆置换(Plasma exchange,PE)治疗,同型病毒灭活血浆2 700ml,置换时间为2.7h,血泵速度:120ml/min,分浆速度:1 000ml/h;隔天1次,共5次;PE完成后患儿头痛逐步消失,颈部疼痛明显缓解、无幻视、无皮肤瘙痒感、四肢肌力5级,复查血清抗AQP4抗体转为阴性,转入普通病房激素和硫唑嘌呤维持治疗。
2 讨论
2021年《中国视神经脊髓炎谱系疾病诊断与治疗指南》(以下简称“指南”)指出以“病史+核心临床症候+影像特征+生物标记物”为基本诊断原则,AQP4-IgG阳性(CBA检测法)患者,需至少满足1个NMOSD核心临床特征,排除其他诊断即可[3]。本例患儿为14岁青春期女性,结合分层诊断标准:血清AQP4-IgG阳性(CBA检测法),同时具有急性脊髓炎、极后区综合征、视神经炎3项核心临床症状,MRI检查符合脊髓炎、视神经炎的影像特点,头部MRI未见脑部病变,且脊髓为延髓至胸8椎体的长阶段损害,排除了多发性硬化(Multiple sclerosis,MS);检测血清中抗髓鞘少突胶质细胞糖蛋白免疫球蛋白G抗体(Anti-myelin oligodendrocyte glycoprotein-IgG, MOG-IgG)阴性,排除儿童发病率高MOGAD,故诊断儿童AQP4-IgG阳性的NMOSD明确,EDSS评8.0分(高分组)。
NMOSD 治疗包括急性期治疗、序贯治疗 (预防复发治疗)、对症治疗和康复治疗,急性期治疗是NMOSD治疗关键环节,以减轻症状、改善致残程度、缩短病程、防治并发症为目的,包括糖皮质激素冲击治疗、静脉注射人免疫球蛋白、血浆置换(Plasma exchange,PE)或免疫吸附治疗(Immunoadsorption,IA)[3]。静脉注射甲泼尼龙可促进神经功能恢复、改善视力,推荐剂量为1g/d,疗程3~5d,后续根据病情及序贯药物起效时间逐渐减量[3-5]。大剂量激素冲击效果不佳者用人免疫球蛋白,对急性期残障功能恢复有益[6],指南推荐静脉滴注人免疫球蛋白0.4g/(kg·d),5d为1个疗程。故该患儿在使用甲泼尼龙冲击治疗无效后积极给予应用1个疗程人免疫球蛋白治疗。指南对于PE适用人群,由先前的“部分NMOSD重症患者尤其是对大剂量甲基泼尼松龙冲击疗法反应差者”,放宽为“中重度发作的NMOSD患者”,免疫吸附治疗无须血浆补充,作为PE一种新型替代疗法,起到类似PE作用机制,推荐有条件单位可以开展,PE/IA升级为A级推荐[3-4],肯定了PE在NMOSD治疗中地位。该患儿在激素冲击治疗及1个疗程人免疫球蛋白后6d,临床呈加重趋势,EDSS评分从8.0分增加至8.5分,提示激素及人免疫球蛋白治疗效果差,考虑在激素维持基础上联合应用PE。
PE是一种血液净化模式,通过血浆分离器将患者血浆进行分离并弃掉,补充外源性血浆,弃浆过程将血浆中大分子物质如免疫球蛋白、白蛋白、致病因子去除,达到治疗疾病目的。PE选择,取决于血液中要清除物质的相对分子量大小、蛋白结合率、分布容积等[5],对于难治性重症自身免疫性疾病常选用血浆置换及集成血液净化模式清除血液中抗原抗体复合物[7-8]。AQP4-IgG是一种免疫球蛋白,相对分子质量约为150kD,属于大分子物质,普通血液透析、血液灌流模式不能将其清除,故应用血浆置换的模式可以将AQP4-IgG清除。
血浆置换对急性发作NMOSD回顾性研究中,分析52例NMOSD患者67次发作事件,对比IVMP和(或)PE治疗的效果,应用扩展残疾状态量表(Extended Disability Status Score,EDSS)和改良Rankin量表(mRS)进行疗效评估,28次发作对单用激素治疗无效,加用PE治疗,随后发现:12次发作对激素联合PE治疗有效,16次发作对激素联合PE仍无反应,研究认为:对于单独使用激素无效NMOSD患者急性发作,应考虑加用PE治疗[9]。2020年关于血浆置换治疗NMOSD时机的荟萃分析,在228例NMOSD患者中应用血浆置换,发现PE治疗后患者EDSS评分平均降低1.04分;在发病7d内进行PE治疗患者,EDSS评分平均下降0.64分。在发病8~23d内进行PE治疗患者,EDSS评分平均下降1.41分。荟萃分析得出结论:PE可改善严重NMOSD,PE开始最佳时间为发病8~23d[10],但是入选的病例全部为成人,未有儿童病例纳入。
本例患儿前期治疗提示激素冲击治疗及人免疫球蛋白治疗无效,于病程第10天开始,在激素维持基础上联合PE,第3次血浆置换(PE)治疗结束后,患儿临床症状明显缓解,5次PE治疗完成,患儿头痛及颈背部疼痛消失、无幻视、无皮肤瘙痒感、四肢肌力达5级,ESDSS评分恢复到7.0分,PE结束后第2天复查血清抗AQP4抗体(CBA检测法)转为阴性。患儿病情达到临床缓解,后续转入普通病房进行维持治疗。2021版指南对于缓解期治疗措施中,规范免疫抑制剂选择、利妥昔单抗(RTX)和小剂量激素使用,增加萨特利珠单抗(Satralizumab),与RTX同为A级推荐[3-4]。本例患儿,因家庭后续经济因素,在小剂量激素维持治疗下,选择口服硫唑嘌呤维持治疗,现随访1个月,病情稳定,未复发。
NMOSD临床表现复杂多样,儿童患者罕见,且以消化道症状首发临床病例更为罕见,极易造成误判,故临床上遇到类似单纯或顽固性呃逆和呕吐病患,对症治疗效果不理想时,应注意NMOSD可能,及时完善该病特异性检验检查协助诊断,及早治疗。血浆置换治疗可能有助于早期清除致病抗体、减轻临床症状、改善致残程度。此为1例儿童个案报道,随访时间尚短,有待更长时间随访及大规模临床研究。
猜你喜欢
现代临床医学(2022年4期)2022-09-29 07:38:00
保健医苑(2022年4期)2022-05-05 06:11:08
猪业科学(2022年2期)2022-04-21 02:18:26
昆明医科大学学报(2021年4期)2021-07-23 01:21:50
昆明医科大学学报(2021年4期)2021-07-23 01:21:32
现代畜牧科技(2021年3期)2021-07-21 08:42:14
中国生殖健康(2020年2期)2021-01-18 02:51:14
中国生殖健康(2019年8期)2019-01-07 01:18:00
海南医学(2016年8期)2016-06-08 05:43:00
磁共振成像(2015年2期)2015-12-23 08:52:22
