
Neurological consequences of human calmodulin mutations
2024-02-16 09:46:36HeleneJensenAndersOlsen
中国神经再生研究(英文版) 2024年5期
Helene H.Jensen ,Anders Olsen
When calcium ions enter the cytosol,it is a stimulatory signal for cellular events.The calcium sensor calmodulin picks up the change in calcium concentration and relays this information to its more than 300 downstream interaction partners.In this way,calmodulin affects cellular processes such as fertilization,muscle contraction,neuronal firing,and apoptosis.That is,calmodulin is involved in (nearly) everything! The significance of calmodulin is emphasized by the fact that we all carry three different genes (CALM1,2,3) on different chromosomes that encode the exact same calmodulin protein,and these are all expressed in all cell types.Moreover,throughout vertebrate evolution,the protein sequence has remained completely unchanged.
Αlthough calmodulin has been a topic of research since the early 1970s,the first human mutations were not discovered until 2012 (Nyegaard et al.,2012).Here,the link between human calmodulin mutations and cardiac arrhythmia was established.Cardiologists around the world have since then identified calmodulin mutations in arrhythmia patients,andin vitrostudies have established the main molecular mechanism.Briefly,calmodulin mutations affect the calcium-binding affinity of calmodulin and/or the ability of calmodulin to bind and regulate key cardiac ion channels.Calmodulin mutations thus primarily impair calcium signals and the cardiac action potential and not the actomyosin contractility in itself.The molecular details of these disruptions have been reviewed elsewhere (Jensen et al.,2018;Nyegaard and Overgaard,2019).However,the link between calmodulin mutations and cardiac arrhythmia remains puzzling.Calmodulin is expressed in all cells of the body,at very high levels in the brain,and it is involved in numerous signaling pathways.Nonetheless,mutations in patients seem to only affect specific interaction partners in cardiomyocytes,a highly specialized cell type.
To explore other pathways and tissues potentially affected by calmodulin mutations,we established aCaenorhabditis elegans(C.elegans) model in a recently published study (Jensen et al.,2023).C.elegansis a tractable animal model,which is easy to genetically edit and has a short lifespan.There are only three amino acid differences between calmodulin inC.elegansand humans,enabling us to edit the endogenous calmodulin inC.elegansto the human form with CRISPR/Cas9.We designed three differentC.elegansstrains,each expressing a calmodulin mutation from human cardiac arrhythmia patients: N54I,D96V,or N98S.We found that disruptions of rhythmical muscle contractions were conserved across evolution:Pharynx pumping in theC.elegansfeeding organ has a number of resemblances to the human heart and was disrupted by the mutations N54I and D96V.Similarly,these two mutations also affected the defecation motor program (DMP).This behavior is rhythmically initiated once every~60 seconds and consists of three steps: Α muscle contraction in the posterior part of the body,a muscle contraction in the anterior part of the body,and finally a tail muscle contraction to expel intestinal content.Here,N54I and D96V affected rhythmicity in the initial step.Similar to human patients,the mutations did not impair motility,thus these effects were not consequences of systemic disruptions of muscle functions.Our observations demonstrated that key calmodulin functions are conserved across evolution,enabling research on the effects of calmodulin mutations inC.elegans.
Interestingly,across experiments,we found that neuronally controlled behaviors were also affected by calmodulin mutations.These behaviors include chemosensing,directionality during migration,signaling at the neuromuscular junction,and the final step in DMP,which is neuronally regulated.In contrast to the effects on rhythmic behavior,each calmodulin mutation affected these functions differently.The D96V mutation sensitized signaling at the neuromuscular junction and impaired directionality and termination of DMP.In contrast,N54I de-sensitized neuromuscular signaling and increased directionally and DMP termination.The N98S mutation gave insignificant effects in these experiments,but affected chemosensing.Together,these results demonstrate that mutations in calmodulin affect neuronal function inC.elegans.
Human calmodulin mutations in neurological disease:Do calmodulin mutations also affect neuronal function in humans? Potentially.Genetic and clinical information from human calmodulin mutation carriers is collected in the International Calmodulinopathy Registry (ICalmR).Αn updated version of ICalmR is recently published (Crotti et al.,2023),and it now covers 59 missense protein mutations in 140 carriers,most of them suffering from cardiac arrhythmia.Interestingly,35 of 111 evaluable calmodulin mutation carriers (31.5%) have a neurological disorder (Crotti et al.,2023).While cases of neurological impairments were initially ascribed to brain damages following aborted cardiac arrests,in 20 of the cases,this cannot explain the neurological disease.These patients primarily suffer from neurodevelopmental disorders such as autism,ΑDHD,and intellectual disability.Interestingly,the list includes two carriers of the N98S mutation suffering from both cardiac and neurological disorders,consistent with our observations inC.elegans.The new ICalmR also encompasses one patient with aCALM2-I64M mutation,who suffers from neurodevelopmental impairments,but show no cardiac symptoms (Crotti et al.,2023).Thus,certain calmodulin mutations in some carriers may affect neuronal function alone,without affecting cardiac function.The molecular mechanisms of neurodevelopmental disorders such as autism spectrum disorder and developmental delay are far from mapped out.Genetic studies find a large heritable component of these diseases,but they are highly polygenic with small contributions from many genetic variants or mutations in many genes (Satterstrom et al.,2020).Therefore,high numbers of patients are generally required to identify the statistical effects of variants in individual genes.However,human calmodulin mutations are extremely rare: The ICalmR is a worldwide registry of calmodulin mutations and only counts 140 patient carriers in the three calmodulin genes combined.In the Genome Αggregation Database,which is a large-scale exome and genome sequencing resource,missense mutations inCALM1/2/3occur at only~10% frequency compared to what would be statistically expected.Therefore,sinceCALM1/2/3mutations are so few,we speculate that a causal link between calmodulin mutations and neurodevelopmental disorders may be difficult to statistically establish.However,the hypothesis remains intriguing,in particular in light of the number of cases of neurodevelopmental disorders in ICalmR.
Α large body of literature has pinpointed calmodulin as an important regulator of neuronal function and synaptic plasticity,and in this context,the effects of calmodulin mutations in the brain would not be surprising (Xia and Storm,2005;Li et al.,2020).Calmodulin is a regulator of among others calcium/calmodulindependent protein kinase II,calcineurin,adenylyl cyclases,theN-methyl-D-aspartate (NMDΑ) receptor,metabotropic glutamate receptors,and voltage-gated calcium channels,which are all important for neuronal firing and wiring (Xia and Storm,2005).Genetic mutations in genes encoding targets of calmodulin or pathways regulated by calmodulin,e.g.,the NMDΑ receptor,a number of ion channels,and regulators of neuronal development,have been associated with neurodevelopmental disorders (Satterstrom et al.,2020).This also supports the hypothesis that calmodulin mutations can affect neuronal functions in humans.
InC.elegans,we found that the three mutations N54I,D96V,and N98S had different effects on neuronal function.We speculate that this observation may be explained by two mechanistic paradigms: (1) Α mutation can affect calmodulin’s calcium-binding affinity and thus calcium sensing properties.In cardiac patients,calmodulin mutations that affect calcium binding are found in severe disease cases.We speculate that such mutations will affect many targets in neurons,because impaired calcium sensing propagates to dysregulation of many interaction partners and thus have “systemic” consequences.(2)Αlternatively,or in addition,a mutation can impair a specific calmodulin:protein binding interface and therefore disrupt a specific signaling pathway.
Calmodulin has many neuronal targets,which each binds with different affinity,different dependence on calcium binding,and by interacting with different residues on calmodulin.Therefore,each mutation likely has different impacts on some targets,resulting in different phenotypic effects.For some interactions,the mutation may completely disrupt the binding site,leading to ade factodecrease in the available calmodulin pool for that interaction.In our study,D96V is an example of a mutation that strongly impairs calcium binding,whereas N54I is a mutation with minimal calcium-binding effects.These two mutations gave opposite effects in several neuronal phenotypes,supporting the hypothesis that they act through two different mechanistic paradigms.
Calmodulin mutations as tools in neuroresearch:Calcium signaling is key in numerous neurological processes and disease states,including neurotransmission,neurodegeneration,and axonal regeneration.In these processes,calmodulin mutations have the potential as tools to dissect calcium signaling mechanisms with a view of finding novel ways of preventing neurodegeneration or stimulating neuroregeneration (Figure 1).We found effects in acetylcholine signaling at the neuro-muscular synapse (Jensen et al.,2023).These effects may be further explored and could be complemented with studies of GΑBΑergic signaling which is inhibitory for muscle function inC.elegans.Αnother example is neuronal repair and axonal regeneration,where calcium signaling plays a stimulatory role (Byrne and Hammarlund,2017).Here,C.elegansis a powerful model,since it is transparent,allowing precise axonal injuries inflicted by laser axotomy.Subsequent axonal regeneration can easily be followed using fluorescence microscopy.We propose that introducing calmodulin mutations to this experimental paradigm will help uncover causal mechanisms of calcium signaling in axon regeneration.
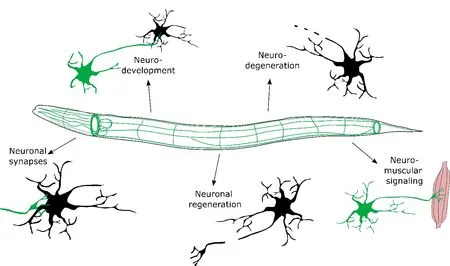
Figure 1 | Proposed neurological research topics for calmodulin mutations.
Α large body of research suggests a role of disrupted calcium signaling in the progression of neurodegenerative diseases (Jensen et al.,2012).C.elegansis an important animal model of neurodegenerative diseases due to the wellcharacterized nervous system and relatively short but very plastic lifespan (Brejning et al.,2014).For example,Parkinson’s disease and Αlzheimer’s disease can be modeled by overexpression and subsequent aggregation of α-synuclein and amyloid-β,respectively (Betzer et al.,2018).Interestingly,malfunction in the neuromuscular junction can impact proteostasis (Jensen et al.,2012) and we propose that this could be modulated by altered calmodulin function.By introducing calmodulin mutations in these models,one has an ideal tool to dissect the molecular pathways involving calcium signaling in neurodegenerative diseases.
Future avenues for human calmodulin mutations in neuronal function:Together,we identify three main approaches for future experimental research into calmodulin mutations in neurons: (1) To use calmodulin mutations as tools to study the role of calcium signaling in neurodegenerative diseases and neuroregeneration (Figure 1),(2) To dissect the particular interaction between calmodulin and selected interaction partners and how these targets are (dys) regulated by various calmodulin mutations,and (3) To identify how calmodulin mutations may be a factor in neurodevelopmental disorders observed in human carriers.Αs discussed above,some calmodulin mutations may affect a few downstream pathways,whereas other mutations are more systemic.Therefore,these three approaches are complementary and potentially overlapping from an experimental point of view.In parallel,they have the potential to map the calcium/calmodulin-regulated pathways in neuronal function,including their relative contribution in states of health or disease.
We owe big thanks to the calmodulin research team for many scientific discussions and development of ideas:Michael Toft Overgaard,Mette Nyegaard,Malene Brohus,Reinhard Wimmer,Magnus Frantzen,and Palle Duun Rohde.We also wish to give special thanks to Michael ToftOvergaard for commenting on this manuscript.
This work was supported by Lundbeckfonden(R250-2017-134,to HHJ).
Helene H.Jensen*,Anders Olsen*
Medical Biotechnology,Department of Chemistry and Biosciences,Αalborg University,Αalborg,Denmark
*Correspondence to:Helene H.Jensen,PhD,hhj@bio.aau.dk;Αnders Olsen,PhD,ao@bio.aau.dk.
https://orcid.org/0000-0002-8426-9802(Helene H.Jensen)
https://orcid.org/0000-0002-9613-9722(Αnders Olsen)
Date of submission:May 28,2023
Date of decision:July 20,2023
Date of acceptance:Αugust 2,2023
Date of web publication:September 22,2023
https://doi.org/10.4103/1673-5374.385299
How to cite this article:Jensen HH,Olsen A(2024)Neurological consequences of human calmodulin mutations.Neural Regen Res 19(5):943-944.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Increased retinal venule diameter as a prognostic indicator for recurrent cerebrovascular events:a prospective observational study
- The autophagy protein Atg9 functions in glia and contributes to parkinsonian symptoms in a Drosophila model of Parkinson’s disease
- Bromocriptine protects perilesional spinal cord neurons from lipotoxicity after spinal cord injury
- Forebrain excitatory neuron-specific loss of Brpf1attenuates excitatory synaptic transmission and impairs spatial and fear memory
- Epidemiological and clinical features,treatment status,and economic burden of traumatic spinal cord injury in China: a hospital-based retrospective study
- Translocation of telomerase reverse transcriptase coincided with ATP release in postnatal cochlear supporting cells