
基于KMP算法的生物序列模式自动识别应用研究
2024-02-09 00:00:00王艳冀松刘静
电脑知识与技术 2024年36期



关键字:KMP算法;生物序列比对;自动识别
中图分类号:TP311 文献标识码:A
文章编号:1009-3044(2024)36-0042-03 开放科学(资源服务) 标识码(OSID) :
0 引言
随着生物科学技术的迅猛发展,生物信息学逐渐在生命科学研究中占据重要地位。生物信息学是生命科学与计算机科学等多学科交叉融合的新兴学科,在基因组学、蛋白质组学等领域发挥着越来越重要的作用。目前,生物信息学已大量应用于生物序列的发现和预测。随着基因组计划的完成和海量生物序列数据的积累,从序列中得到的信息已经比迄今为止的所有生物研究积累的信息还要多,而如何高效地识别和分析生物序列模式成为亟待解决的关键问题。
生物序列分析是生物信息学的核心内容之一,而高效的序列模式识别算法对于从海量生物数据中提取有用信息至关重要。本文将从对生物序列模式和KMP算法的研究开始,通过分析生物序列和字符序列的相似性,将复杂的生物序列分解为对应的一段段的字符序列,进而采用字符串的模式匹配算法来解决生物序列的比较问题,实现生物序列模式的自动识别。
1 生物序列模式相关概念
生物序列是指由核苷酸或氨基酸组成的线性序列,携带着重要的遗传信息或蛋白质结构信息。其中,核苷酸共有四种,分别为腺嘌呤、胞嘧啶、鸟嘌呤、胸腺嘧啶[1]。由核苷酸聚合而成的生物序列称为核酸序列,又分为脱氧核糖核酸(deoxyribonucleic acid,DNA) 和核糖核酸(ribonucleic acid,RNA) 两种[2]。核酸序列携带着重要的遗传信息,该遗传信息与核苷酸的排列顺序有关;氨基酸共有20种,由氨基酸连接而成的生物序列称为蛋白质序列,蛋白质的结构与氨基酸的排列次序有关。
生物序列模式是指在DNA、RNA或蛋白质序列中重复出现的特定序列或结构特征。这些模式可能在一组相关的蛋白质、DNA或RNA序列中反复出现,代表着生物系统中非常重要的遗传信息或结构信息,通常具有特定的生物学功能。
随着人类基因组计划工程的完成,其他生物基因组的研究也取得了突破性进展。某些生物,如酵母菌、果蝇、线虫等,具有繁殖能力强、生长快、结构简单等特点。它们的基因组结构和功能已经被广泛研究、了解并应用。
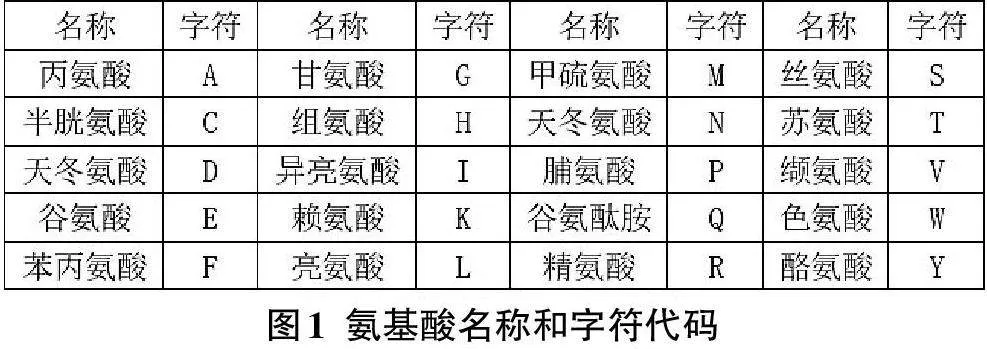
核酸序列共由四种核苷酸组成,分别由A(腺嘌呤) 、T(胞嘧啶) 、C(鸟嘌呤) 、G(胸腺嘧啶) 四个字符表示;蛋白质序列由二十种氨基酸组成,其对应的字符代码如图1所示。因此,生物序列比对可以转化为字符串比对问题。
2 模式匹配算法基本原理
字符串匹配算法在生物信息学、信息检索等领域具有广泛的应用。所谓字符串匹配算法,是指假设存在两个字符串S和T,其中字符串S的长度为n,字符串T的长度为m。若要查找串T是否是串S的子串,则称串S为目标串,将T称为模式串,把查找模式串T在目标串S中的匹配位置的运算称为模式匹配。如果在目标串S中查找到一个与模式串T相同的字符串,则模式串与目标串匹配;如果在目标串S中未查找到一个与模式串T相同的字符串,则不匹配。本文重点介绍BF算法和KMP算法。
2.1 传统的模式匹配算法
传统的模式匹配算法即BF(Brute Force) 算法[3],亦称简单匹配算法,采用穷举的思路,穷举目标串S的所有子串,判断是否与模式串T匹配。算法的基本思想是从目标串S的某个字符开始,依次与模式串T的每一个字符进行匹配。若相等,则继续比较下一个字符;若不相等,则匹配不成功,目标串S回退。下一次比较时,模式串T回退到首字符,目标串S只右移一个字符,直到模式串T中的每个字符依次与目标串S中的一个连续字符序列相等,则匹配成功。BF算法时间复杂度高,平均时间复杂度为O(n×m)。因此BF算法的匹配过程虽易于理解,但效率一般。
2.2 模式匹配的改进算法
模式匹配的改进算法即KMP(Knuth-Morris- Pratt) 算法,它充分利用已经部分匹配的结果,尽量避免不必要的字符比较,改进了造成BF算法速度慢的回溯特点。在某次匹配过程失败后,指向目标串S的指针不回溯,而是让模式串T向右移动一定的距离,然后继续与目标串S该位置的字符对齐,依次比较,直到匹配成功为止。整个匹配过程中,只需要对目标串从头到尾扫描一遍,指向目标串的指针不需要回溯,模式串也无须从头开始。KMP算法具有线性时间复杂度O(n+m),与BF算法相比,大大减少了比较次数,提高了匹配效率。KMP算法比较适合模式串较长并多次出现在目标串中的情况,例如在文本编辑器中的查找和替换功能、DNA的序列匹配等,都可以通过采用KMP算法提高匹配效率。
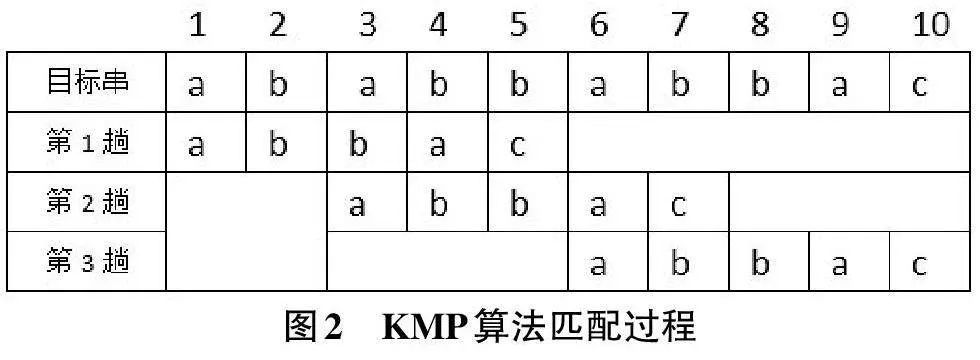
下面通过一个例子来说明KMP算法的匹配过程。例如,目标串S为“ababbabbac”,模式串T为“abbac”,按照KMP算法进行匹配。
第1趟匹配过程中,目标串S从第1个字符开始,模式串T的第1个字符与其对齐,依次向右进行匹配。比较到目标串的第3个字符时第一次出现不匹配,模式串T向右移动两个字符;第2趟匹配过程中,目标串S从第3个字符开始,模式串T的第1个字符与其对齐,依次向右进行匹配。比较到目标串的第7个字符时第一次出现不匹配,模式串T向右移动三个字符;第3趟匹配过程中,目标串S从第7个字符开始,模式串T的第2个字符与其对齐,依次向右进行匹配,最终匹配成功。具体匹配过程如图2所示[4]。
3 KMP 算法设计与实现
根据以上分析可知,KMP算法在匹配失败时,能快速定位到下一个可能的匹配位置,解决模式串向右移动的最大距离问题是关键。
定义目标串S和模式串T,设指针i和j分别指向S 和T中正待比较的字符Si和Tj。根据KMP算法的思想[5],当Si不等于Tj时,目标串S的指针i不动,模式串T向右移动。此时,模式串T的前j-1个字符已经与目标串S 的前i-1 个字符相匹配,即“Si-j+1…Si-1”==“T1…Tj-1”(1) 。假设模式串向右最多移动到Tk时,Si==Tk,并且有“Si-k+1…Si-1Si”==“T1…Tk-1Tk”(2) ,此时应继续比较下一个字符Si+1和Tk+1。因为k 必然小于j,由(1) 等式可以得到部分匹配“Si-k+1… Si-1”==“Tj-k+1…Tj-1”(3) ,由(2) 和(3) 两个等式可以推出“T1…Tk-1”==“Tj-k+1…Tj-1”(4) ,其中“Tj-k+1 T…kT-j1-”1表”表示示模模式式串串T中Tj前的的相同最字长符后的缀前子缀串子,而串“。T1…因此,当目标串Si与模式串Tj不匹配时,模式串T向右移动的距离仅与模式串T自身有关,而与目标串S无关。为此,在KMP算法中引入数组next[],用next[j]的值表示这个最长后缀子串的长度,核心问题是求解数组next[j]的值。
3.1 next[]数组
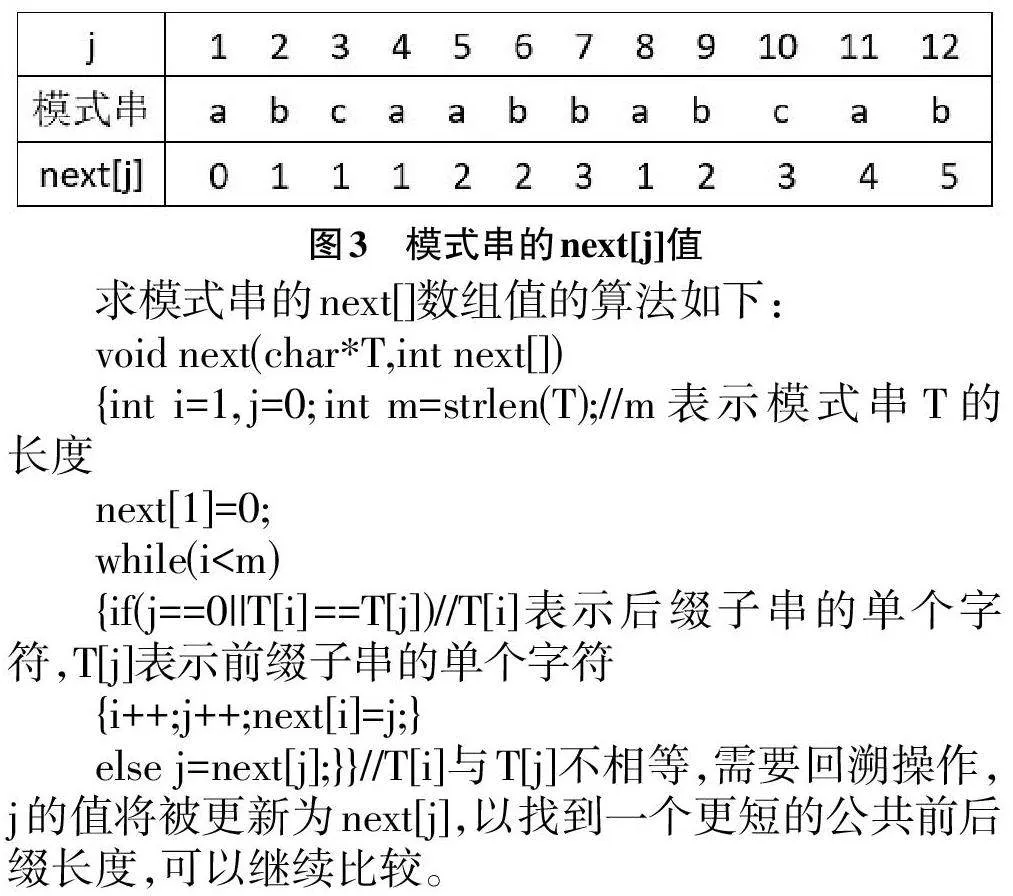
由以上分析可知,next[]数组是针对模式串T构建的一个辅助数组,其长度与模式串T 的长度相同。next[j]用于表示模式串T中第j个字符位置之前的子串(从模式串T的第一个字符开始到第j个字符的子串) 的最长公共前后缀长度。
由于模式串T的第一个字符前没有子串,因此有next[1]=0。用指针i遍历模式串T,比较模式串T的第i 个字符和第j个字符(即Ti和Tj) 。如果Ti等于Tj,则说明当前位置的子串的最长公共前后缀长度可以在前一个位置(i-1) 的基础上增加1,即next[i]=next[j]+1,然后i和j都向后移动一位,继续判断下一个位置。如果Ti不等于Tj,则可将求next[j]值的问题继续看作一个模式匹配的问题,整个模式串既是目标串又是模式串,查找最长公共前后缀子串。若没有公共前后缀子串,则next[j]=1。
3.2 KMP算法实现
在已经求得模式串的部分匹配结果即next[]数组值的前提下,进行KMP算法实现。
4 KMP 算法在生物序列模式自动识别中的应用
随着基因组计划和大量模式基因组工程的完成,目前已经建立了与基因组信息相关的数据库,绝大部分的生物序列也根据其不同的生物学意义采用不同的数据库进行存放。由于生物序列通常很长,并存在许多具有特定功能的模式,自动识别这些模式对于理解基因的结构和功能、基因表达调控以及疾病相关基因的发现等方面具有极其重要的意义。例如,当研究人员研究一种新出现的病毒时,通过基因组计划所提供的信息,可能会找到某个或某些相关基因,从而较快速地确定病毒序列的结构和特点。这就需要在这样长的生物序列中查找特定的模式序列,找出具有相似性的同源序列。KMP算法与无回溯的线性时间复杂度相比BF算法具有更高的效率,能够在相对较短的时间内处理大量的生物序列数据。KMP算法在生物序列模式自动识别中的应用可以分为预处理和匹配识别两个阶段。
4.1 预处理阶段
首先,根据要识别的生物序列模式特征,组合构建对应的模式序列。将构建好的模式序列作为模式串,将要查找的基因主序列作为目标串,分别转化为对应的字符序列。然后,根据模式串计算其部分匹配表(next[]数组) ,在后续的匹配过程中用于快速跳过已经匹配过的部分,从而提高匹配效率。
4.2 匹配识别阶段
将模式序列与基因主序列进行匹配, 当模式序列完全匹配基因主序列中的某一段时,就确定了基因模式的位置。具体实现过程在KMP算法基础上做了进一步扩展,当匹配成功后不停止搜索,而是继续在主序列中查找直到主序列遍历完为止,实现了在主序列中查找模式序列的所有位置并输出。当查找成功时将匹配位置记录在数组indices 中,同时将j 更新为next[j-1],以继续搜索下一个可能的匹配。以下为匹配过程的核心代码部分:
以上为KMP算法在生物序列模式自动识别中的应用的具体实现,该算法可以应用于疾病诊断、微生物鉴定、生物进化研究等多个生物学方向。例如,在微生物鉴定方面,针对已知的致病性微生物基因序列模式,使用自动识别算法在未知微生物的基因组序列中进行搜索,可以快速鉴定微生物是否为致病性菌株。以识别大肠杆菌的志贺毒素基因(stx) 为例,将stx 基因序列模式作为模式序列,将从临床样本中分离出的大肠杆菌基因组序列作为目标序列进行匹配。如果识别出stx基因序列,那么就可以初步判定该大肠杆菌菌株具有致病性,这对于及时采取治疗措施和公共卫生防控具有重要意义。
5 结束语
综上所述,本文在介绍生物序列模式和模式匹配算法的基础上,通过将生物序列转化为字符序列,实现了KMP算法在生物序列自动识别中的应用。该算法能够高效且准确地识别生物序列中的序列模式,为生物信息学研究提供了重要的技术支持。未来,随着生物序列数据量的持续增长,KMP算法还可与其他先进的生物信息学技术深度融合,不断提高处理大规模生物序列数据时的效率和准确性,进一步推动生物信息学的发展。
猜你喜欢
电子制作(2019年13期)2020-01-14 03:15:32
移动信息(2018年1期)2018-12-28 18:22:52
中央民族大学学报(自然科学版)(2018年3期)2018-11-09 01:16:38
特别健康(2018年3期)2018-07-04 00:40:18
发明与创新(2016年26期)2016-08-22 03:23:28
电测与仪表(2016年6期)2016-04-11 12:06:38
山东工业技术(2015年21期)2015-07-27 08:18:10
电脑迷(2014年8期)2014-04-29 07:37:40
计测技术(2014年6期)2014-03-11 16:22:12
卷宗(2011年9期)2011-05-14 17:51:19
