一测多评法同时测定化肝煎中5种成分及其质量评价*
2024-01-25 10:55罗曼张武岗喻文韬吴欢姚闽冯育林杨世林
现代中医药 2024年1期
罗曼 张武岗, 喻文韬 吴欢 姚闽,4 冯育林, 杨世林**
(1.江西中医药大学,江西 南昌 330006;2.中药固体制剂制造技术国家工程研究中心,江西 南昌 330006;3.江西本草天工科技有限责任公司,江西 南昌 330006;4.江西省药品检验检测研究院,江西 南昌 330006)
化肝煎由青皮、陈皮、白芍、牡丹皮、栀子(炒)、泽泻、贝母组成,最早记载于明代张景岳的《景岳全书》[1],2018年4月被收载入《古代经典名方目录(第一批)》[2]。目前多用于治疗因肝气上逆致肝胃不和引起的消化系统疾病和妇科类疾病[3-7]。目前,关于化肝煎的多成分质量控制方法研究报道较少,聂欣等[8]建立了化肝煎中芍药苷、橙皮苷和丹皮酚3种成分含量分别测定的HPLC方法;杨雪颖等[3]通过建立2种HPLC方法来分别控制化肝煎中栀子苷、丹皮酚和芍药苷3种成分的含量;郭中华等[9]建立了化肝煎中没食子酸、栀子苷、芍药苷、芸香柚皮苷、橙皮苷5种成分含量的HPLC测定方法。为了更加简便、快捷的对化肝煎的质量进行评价,本研究拟建立化肝煎多成分质量控制的QAMS法,以期为化肝煎的后续开发及质量评价提供参考。
QAMS法是以内标法、外标法(External Standard Method,ESM)、校正因子法等分析方法为基础的延伸,该方法以药物本身所具有的成分为参照,同步进行多个成分含量的测定,不仅解决了在质量控制时,对照品价高和难得的问题,还能更加准确地反映整个处方的质量状态[10-13]。因此,本研究拟以Nari作为参照,建立化肝煎中Gal、Geni、Nari、Hesp和Paeo 5种成分含量的QAMS法,同时利用化学模式识别分析方法对化肝煎不同批次样品的检测结果进行评价[14],以期为该处方的后续开发及质量评价提供一种更为简便的方法。
1 材料
1.1仪器 LC-20AD高效液相色谱仪(日本岛津);JM-B50002电子天平(余姚市纪铭称重校验设备有限公司); MS205DU电子分析天平(METTLER TOLEDO科技有限公司);康雅顺电陶炉(康雅顺电器有限公司);KQ5200DE型数控超声波清洗器(昆山市超声仪器有限公司);HWS-24型电热恒温水浴锅(北京科伟永兴仪器有限公司)。
1.2试剂与试药 陈皮(批号:20201101-5,江西南丰)、青皮(批号:20190601-5,江西吉安)、白芍(批号:201907011-5,浙江磬安)、牡丹皮(批号:1909011-5,四川苍溪)、栀子(批号:20201111-5,江西)、泽泻(批号:20200311-5,福建南平)、浙贝母(批号:20200311-5,浙江磬安县)各5批;以上七味药经江西省药品检验检测研究院高级工程师姚闽鉴定。
橙皮苷(批号:110721-202019,质量分数:95.3%),没食子酸(批号:110831-201906,质量分数:91.5%),栀子苷(批号:110749-201919,质量分数:97.1%),丹皮酚(批号:110708-201908,质量分数:99.8%),以上对照品均购自于中国食品药品检定研究院;芸香柚皮苷(批号:YY-071-190329,质量分数:98%),购自成都瑞芬思生物科技有限公司;甲醇(色谱级,Fisher);磷酸(色谱级,Fisher);纯净水(杭州娃哈哈集团有限公司)。
2 方法与结果
2.1溶液制备
2.1.1对照品溶液 精密称取Gal、Geni、Nari、Hesp和Paeo,以甲醇为溶剂配制质量浓度分别为0.1601、2.1900、1.8030、0.7205和0.1616 mg·mL-1的对照品溶液。同时配制一份Gal、Geni、Nari、Hesp和Paeo质量浓度分别为0.0021、0.0219、0.0060、0.0068和0.0030 mg·mL-1的混合对照品溶液。
2.1.2供试品溶液 称取青皮(20190605)7.50 g,陈皮(20201101)7.50 g,白芍(201907015)7.50 g,牡丹皮(1909013)5.63 g,栀子(20201112)5.63 g,泽泻(20200311)5.63 g,浙贝母(20200714)10.00 g,置煎药壶中,加水300 mL,浸泡30 min,以武火煮沸,转文火煎煮40 min,趁热过滤,即得标准煎液。精密吸取标准煎液10 mL于50 mL容量瓶内,加甲醇定容至刻度线,即得供试品溶液。
2.1.3阴性样品溶液 参照“2.1.2”项分别制备缺青皮;缺陈皮;缺栀子;缺牡丹皮;缺青皮和陈皮;缺青皮、陈皮、牡丹皮和栀子;缺青皮、陈皮、牡丹皮、栀子和白芍的阴性样品溶液。
2.2色谱条件 色谱柱:Welch C18色谱柱(250 mm×4.6 mm,5 μm);流动相:甲醇-0.1%的磷酸水,洗脱时间程序如表1所示;柱温40 ℃;流速1.0 mL·min-1;检测波长270 nm;进样量10 μL。

表1 HPLC梯度洗脱表
2.3方法学考察
2.3.1专属性实验 取“2.1.1”项下混合对照品、“2.1.2”项下供试品溶液和“2.1.3”项下七种阴性样品溶液各适量,按“2.2”项的色谱条件进样测定,结果如图1所示,5种成分色谱峰的分离度均大于1.5,且阴性样品对Gal、Geni、Nari、Hesp和Paeo 5种成分的含量测定没有干扰,表明该方法专属性良好。
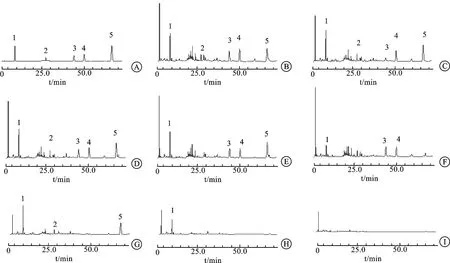
注:A.对照品溶液;B.供试品溶液;C.缺青皮阴性样品;D.缺陈皮阴性样品;E.缺栀子阴性样品;F.缺牡丹皮阴性样品;G.缺青皮、陈皮阴性样品;H.缺青皮、陈皮、栀子和牡丹皮阴性样品;I.缺青皮、陈皮、栀子、牡丹皮和白芍阴性样品;1.Gal;2.Geni;3.Nari;4.Hesp;5.Paeo
2.3.2线性关系考察 将“2.1.1”项下对照品溶液分别稀释成5个浓度梯度,按“2.2”项的色谱条件分别进样测定。横坐标(X)为各成分质量浓度(mg·mL-1),纵坐标(Y)为各成分峰面积,得到各成分的回归方程和线性范围如表2所示,Gal、Geni、Nari、Hesp和Paeo分别在各自的范围内线性关系良好,R2均在0.9992~0.9999之间。

表2 各成分线性关系与范围
2.3.3精密度实验 将“2.1.1”项下混合对照品溶液按“2.2”项的色谱条件连续进样六次,测得Gal、Geni、Nari、Hesp和Paeo含量的相对标准偏差(RSD)值分别为0.25%、0.23%、0.22%、0.30%和0.30%,均小于1.00%,表明该仪器精密度良好。
2.3.4重复性实验 按“2.1.2”项下的方法平行制备同批次6份供试品溶液,分别按“2.2”项的色谱条件进样测定,测得Gal、Geni、Nari、Hesp和Paeo含量的RSD值分别为2.27%、1.77%、0.29%、0.57%和0.34%,均小于3.00%,表明该方法重复性良好。
2.3.5稳定性实验 吸取同一份供试品溶液,按“2.2”项的色谱条件,分别于0、4、8、12、16、24 h进样测定。测得Gal、Geni、Nari、Hesp和Paeo含量的RSD值分别为0.25%、2.19%、0.33%、0.50%和0.33%,均小于3.00%,表明该供试品溶液在24 h内稳定。
2.3.6加样回收实验 参照“2.1.2”项下方法制备两份供试品溶液,精密量取6份标准汤剂,每份2 mL,分别以1∶0.8、1∶1、1∶1.2的比例加入对照品粉末,加入适量甲醇,超声使其完全溶解,冷却至室温,甲醇定容至10 mL,取上清液,过0.22 μm有机滤头,按“2.2”项的色谱条件进样测定,测各成分加样回收率和RSD值。结果测得在加样80%时,Gal、Geni、Nari、Hesp和Paeo的平均加样回收率分别为96.86%、94.29%、104.0%、93.69%和92.77%;加样100%时,Gal、Geni、Nari、Hesp和Paeo的平均加样回收率分别为94.71%、97.53%、105.90%、97.20%和95.04%;加样120%时,Gal、Geni、Nari、Hesp和Paeo的平均加样回收率分别为95.46%、92.87%、101.23%、105.8%和97.28%;加样回收实验的RSD值均小于3.00%,表明该方法准确可靠。
2.4QAMS法分析
2.4.1相对校正因子测定 相对校正因子(fk/s)计算公式为fk/s=fk/fs=(CkAs)/(CsAk),其中C表示质量浓度,A表示峰面积,S表示参照物,K表示其他成分[11-12,15-18]。精密吸取6份“2.1.1”项下混合对照品溶液,按“2.2”项的色谱条件进样测定,以Nari为参照,计算其他4种成分的相对校正因子,结果见表3。
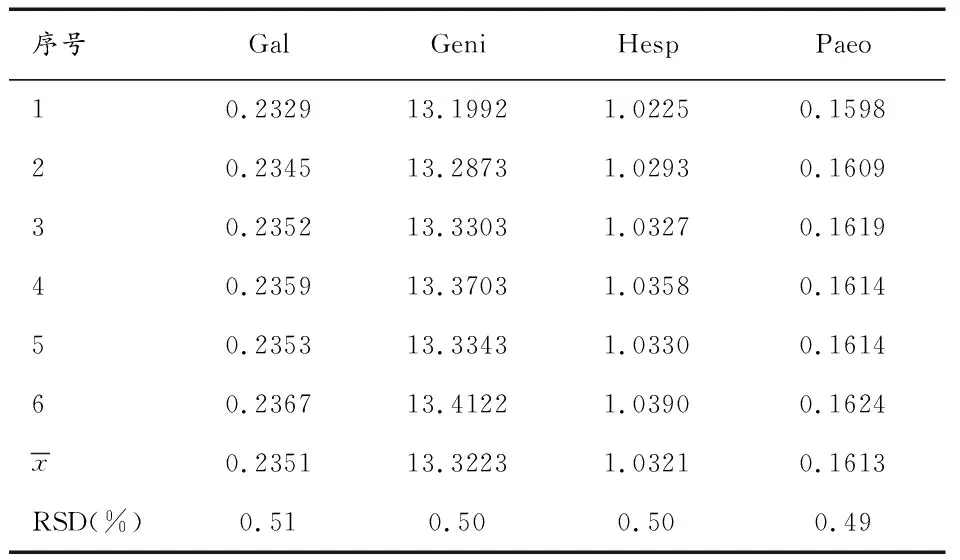
表3 各成分的相对校正因子(Nari为参照)
2.4.2不同色谱柱对校正因子的影响 取“2.1.1”项下的对照品溶液,以Nari为参照,在岛津色谱仪下考察Welch C18、Diamonsil C18和Cosmosl C18色谱柱对相对校正因子的影响,结果如表4所示,这三种色谱柱对相对校正因子的影响不显著(RSD<2.00%)。

表4 不同色谱柱对相对校正因子的影响
2.4.3流动相pH对校正因子的影响 取“2.1.1”项下的对照品溶液,以Nari为参照,在岛津色谱仪、Welch C18(4.6 mm×250 mm,5 μm)色谱柱下,考察不同pH(2.1、2.4、2.7)的流动相对相对校正因子的影响[17-18],结果见表5,从表可知不同pH对相对校正因子的影响不显著(RSD<1.00%)。

表5 不同pH对相对校正因子的影响
2.4.4不同柱温对校正因子的影响 取“2.1.1”项下的对照品溶液,以Nari为参照,在岛津色谱仪、Welch C18 (4.6 mm×250 mm,5 μm)色谱柱下,考察不同柱温(35℃、37℃、40℃、42℃、45℃)对Gal、Geni、Hesp和Paeo 4种成分相对校正因子的影响。结果如表6所示,以Nari为内标物时,不同柱温对这4种成分的相对校正因子的影响不显著(RSD<1.00%)。
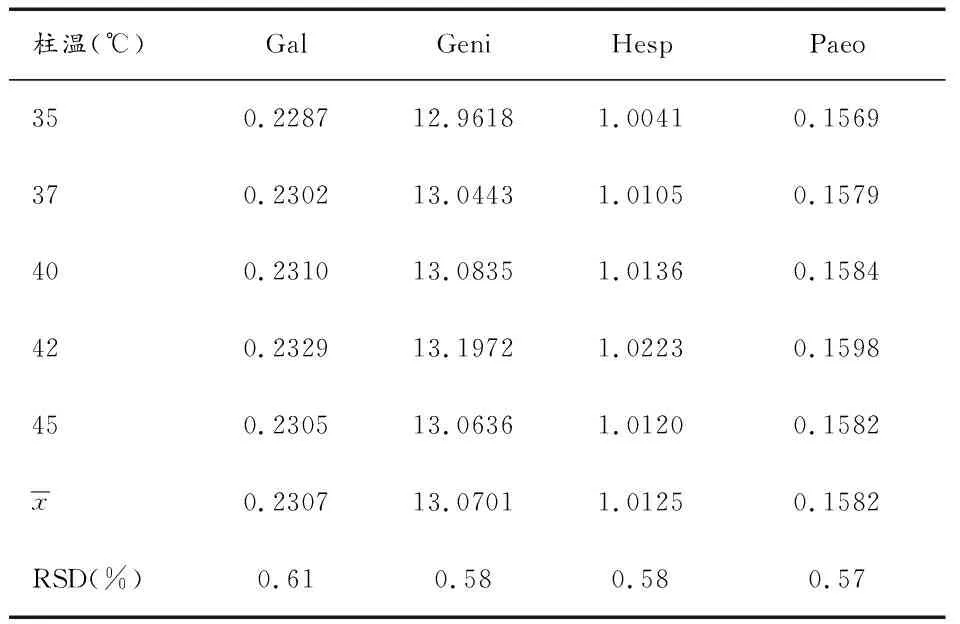
表6 不同柱温对相对校正因子的影响
2.4.5色谱峰定位 相对保留时间计算公式tri/s= tRi/tRs[19],tri/s表示相对保留时间,tRi表示其他成分保留时间,tRs表示内标物保留时间,考察其在Welch C18、Diamonsil C18和COSMOSL C18三种不同色谱柱下的重复性。取“2.1.1”项的混合对照品溶液,用岛津高效液相色谱仪进样测定,以Nari为参照,计算其他四种物质的tri/s和RSD。结果如表7所示,采用该方法所计算的各成分相对保留时间的RSD均小于1.50%,即说明该方法适用。
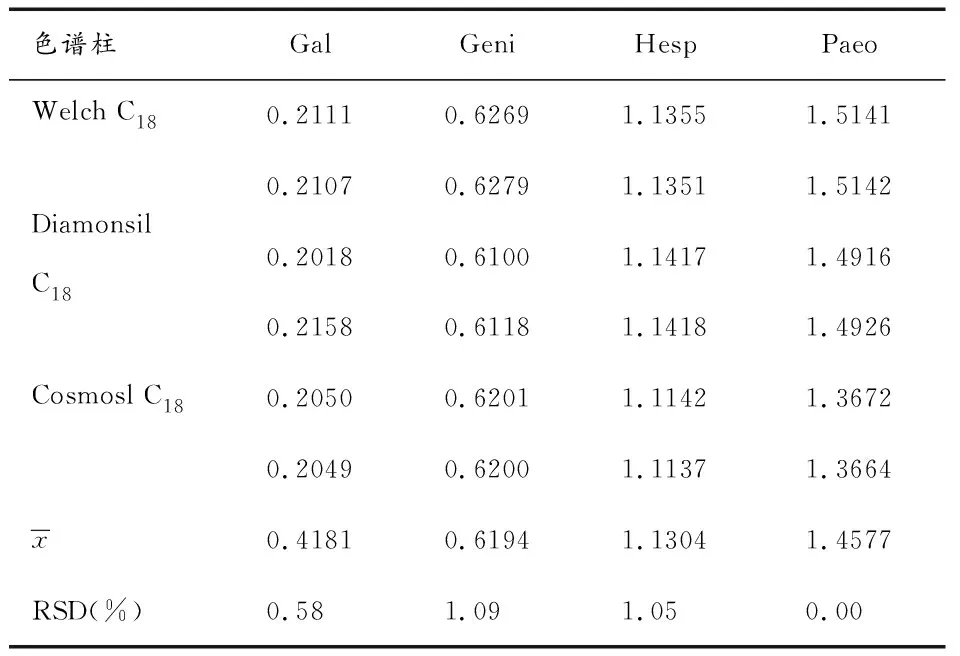
表7 不同色谱柱各成分的相对保留时间值
2.5样品含量检测 将5批次化肝煎7味药材随机组合成15批处方,按照“2.1.2”项制备方法制得15批供试品溶液(编号S1~S15),按“2.2”项的色谱条件分别采用外标法和一测多评法计算各成分含量,结果如表8所示,两种方法所测Gal、Geni、Hesp和Paeo 4种成分含量的RSD值均小于3.00%,说明两种方法测得的各成分含量差异不显著,QAMS法可靠。同时,测得15批次化肝煎中5种成分的含量均未出现离散值,说明供试品的制备工艺稳定可靠。

表8 15 批次样品中各成分含量检测结果(mg·mL-1)
2.6化肝煎化学模式识别分析方法的建立
2.6.1聚类分析 以Nari为参照,将2.5项下15批化肝煎中5种待测成分的QAMS法含量数据导入SPSS 21软件进行聚类分析,结果显示,当欧氏距离为15时,15批样品可分为2大类,S2~S4、S6、S10~S12为一类,S1、S5、S7~S9、S13~S15为一类,其可以评估15批样品之间的差异。
2.6.2主成分分析(PCA) 以Nari为参照,将2.5项下15批化肝煎中5种待测成分的QAMS法含量数据导入SIMCA 14.1软件进行PCA分析,在一定程度上可以进一步分析不同批次化肝煎之间的质量差异,并初步分析出造成该差异的成分。PCA模型自动拟合选择两个主成分,其累计方差贡献率为95%(大于85%),表明模型预测良好[14],结果见图2,15批样品的分组结果与聚类分析一致,S2~S4、S6、S10~S12为一组,S1、S5、S7~S9、S13~S15为第二组。
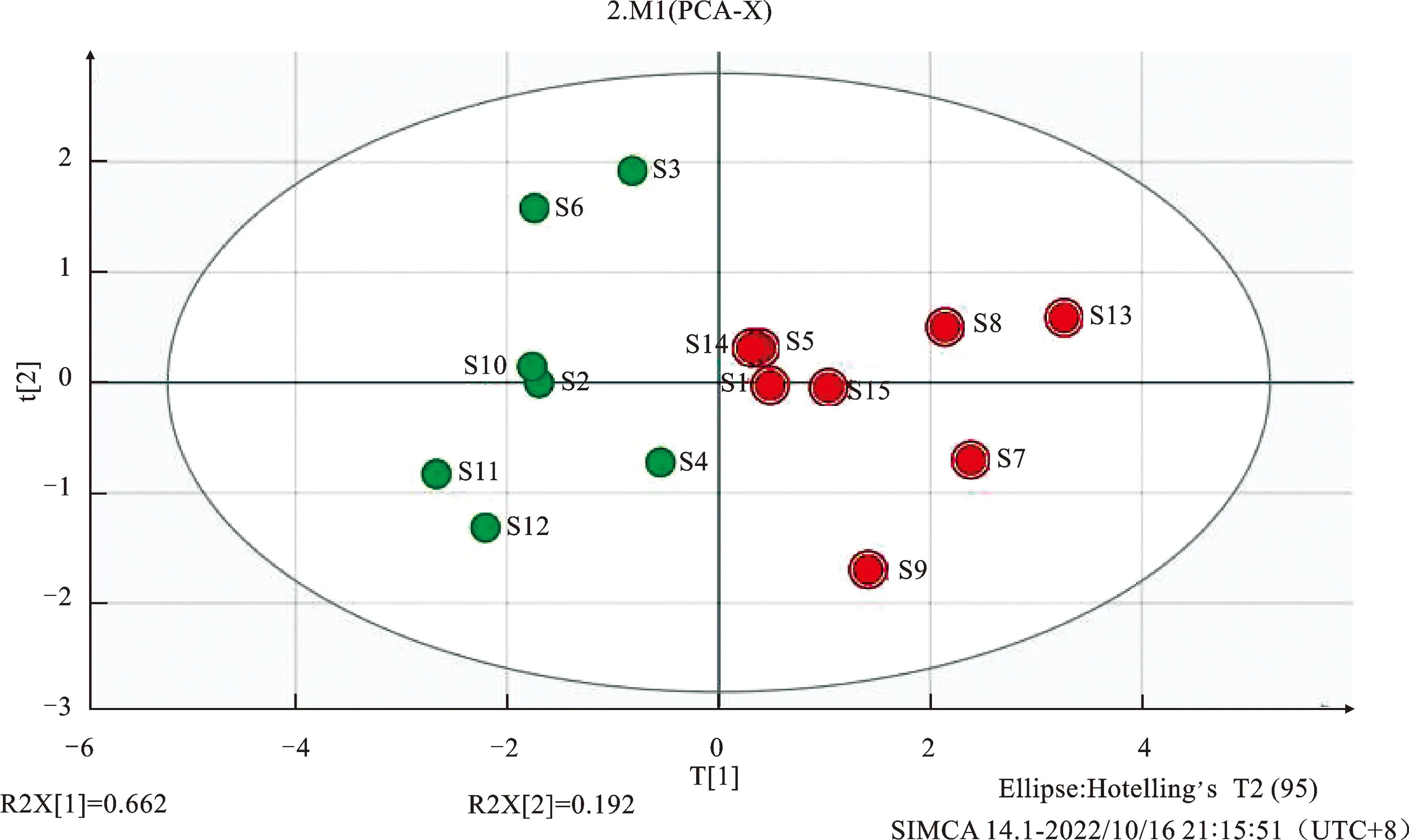
图2 15批化肝煎样品PCA分布图
2.6.3偏最小二乘法-判别分析(PLS-DA) 以“2.5”项下15批化肝煎中5个待测成分的QAMS法含量为变量,通过SIMCA 14.1软件进行PLS-DA分析,然后对PLS-DA模型进行200次置换检验,结果如图3所示。
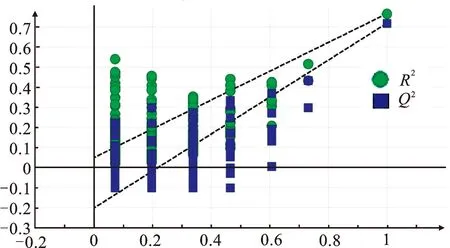
图3 15批化肝煎样品的PLS-DA模型置换检验图
由图3可知:拟合直线Y轴的截距为0.0489<0.3,表明所建立的模型结果可靠;Q2拟合直线Y轴截距为-0.2020<0.0500,说明所建立的模型没有过度拟合,此分析可有效判别分析15批化肝煎的质量差异。再根据变量重要性投影(variable importance in projection,VIP)分析不同批次样本中相同成分的影响强度,筛选影响化肝煎质量的标志性成分[13],结果显示(图4):Gal、Geni、Nari和Hesp的VIP值均大于1.00,则这4种成分可能是影响该药质量的标志性成分。
3 讨论
3.1色谱条件的选择 本实验分别考察了不同流动相、流速、柱温、检测波长下化肝煎中5种成分在色谱图中的分离度、保留时间、峰数量,实验发现这5种成分在230 nm和270 nm波长下吸收较好,但在230 nm波长下的分离效果不理想;甲醇选择性虽然弱于乙腈,但甲醇溶剂下峰形更优,杂质沉淀更为完全,0.1%的磷酸水的加入使化肝煎中这5种成分的出峰时间更合适[20],峰形较好;流速在0.9 mL·min-1以上时各成分出峰时间和峰形都比较好,1.00 mL·min-1的峰形最好,所以最终选择甲醇和0.1%的磷酸水为流动相、柱温40℃、流速1.00 mL·min-1、进样量10 μL、检测波长270 nm作为检测条件,同时采用梯度洗脱对各成分进行分离和检测。
3.2检测指标的选择 化肝煎具有疏肝理气、泻热和胃之功[21-22]。在指标成分选择时,根据处方君臣佐使的顺序依次进行考虑[23]。青皮和陈皮为君药[24],其中主要含有Nari和Hesp等成分[25-26]。栀子、牡丹皮和白芍为臣药[24],栀子中主要含有Geni[27];牡丹皮中主要含有Paeo和Gal[28];白芍中主要含有芍药苷和Gal等成分[29];但芍药苷色谱峰的分离度较差,难以和其他物质分离开来,故而本实验没有选择芍药苷作为检测指标。泽泻和浙贝母为佐药[24],所含的主要成分色谱峰吸收较差,故本实验未对其进行检测。同时Gal、Geni、Nari、Hesp和Paeo与化肝煎的药理活性均存在直接相关性[19],进而最终将这5种成分定为检测指标。
3.3内标物的选择 在实验中发现,Nari性质稳定,线性拟合度较好,色谱峰分离度合适,峰形易辨认,含量也比较高,对照品价格相对比较便宜。所以根据内标物的选择原则[30-31],本实验选择Nari作为参照。
4 结论
本实验采用QAMS法同时测定化肝煎中Gal、Geni、Nari、Hesp和Paeo的含量,测得这5种成分在各自线性范围内线性关系良好(R2≥0.999),平均加样回收率在92%~106%之间,RSD<3.00%;方法的专属性、精密度、重复性和稳定性良好。Gal、Geni、Hesp和Paeo相对于Nari的校正因子分别是0.2351、13.3223、1.0321、0.1613。QAMS法含量检测结果与外标法无统计学差异,表明本研究建立的QAMS法较为合理。从聚类分析、主成分分析和偏最小二乘法-判别分析结果可以看出,Gal、Geni、Nari和Hesp是影响化肝煎质量的标志性成分。
综上所述,本研究所建立的一测多评法和化学模式识别分析可用于化肝煎的质量评价,且该方法简单便捷,稳定性和重复性好,可为该方的质量控制和评价提供更为全面科学的参考。
猜你喜欢
色谱(2022年11期)2022-11-10
色谱(2022年10期)2022-10-13
色谱(2022年7期)2022-06-25
色谱(2022年4期)2022-04-01
国学(2020年1期)2020-06-29
基层中医药(2018年7期)2018-12-06
中国现代中药(2018年11期)2018-11-19
数学物理学报(2017年6期)2018-01-22
摄影之友(影像视觉)(2017年1期)2017-07-18
金色年华(2016年11期)2016-02-28