
Comparative proteomics reveals the response and adaptation mechanisms of white Hypsizygus marmoreus against the biological stress caused by Penicillium
2024-01-24 01:12XiuqingYangSizhuLiXiaohangLiChenxiaoZhangMeijieLiuLizhongGuoLinLiuHaoYu
食品科学与人类健康(英文) 2024年3期
Xiuqing Yang, Sizhu Li, Xiaohang Li, Chenxiao Zhang, Meijie Liu, Lizhong Guo, Lin Liu, Hao Yu
Shandong Provincial Key Laboratory of Applied Mycology, School of Life Sciences, Qingdao Agricultural University, Qingdao 266109, China
Keywords: Hypsizygus marmoreus Penicillium Proteomics Biological stress response Adaptation
ABSTRACT White Hypsizygus marmoreus is a popular edible mushroom. Its mycelium is easy to be contaminated by Penicillium, which leads to a decrease in its quality and yield. Penicillium could compete for limited space and nutrients through rapid growth and produce a variety of harmful gases, such as benzene, aldehydes,phenols, etc., to inhibit the growth of H. marmoreus mycelium. A series of changes occurred in H. marmoreus proteome after contamination when detected by the label-free tandem mass spectrometry (MS/MS) technique.Some protein s with up-regulated expression worked together to participate in some processes, such as the non-toxic transformation of harmful gases, glutathione metabolism, histone modif ication, nucleotide excision repair, clearing misfolded proteins, and synthesizing glutamine, which were mainly used in response to biological stress. The proteins with down-regulated expression are mainly related to the processes of ribosome function, protein processing, spliceosome, carbon metabolism, glycolysis, and gluconeogenesis. The reduction in the function of these proteins affected the production of the cell components, which might be an adjustment to adapt to growth retardation. This study further enhanced the understanding of the biological stress response and the growth restriction adaptation mechanisms in edible fungi. It also provided a theoretical basis for protein function exploration and edible mushroom food safety research.
1. Introduction
The whiteHypsizygus marmoreusis also known as the white beech mushroom or the crab-flavored mushroom. It is a variant of the greyH. marmoreusstrain taxonomically belonging to the basidiomycetes. It is widely planted in East Asia and Northern Europe.It has been one of the most popular edible fungi loved for its unique soil flavor, chewy texture and rich taste, excellent nutrition, and medicinal value[1-2]. The fruiting body of the whiteH. marmoreushas high protein content and rich mineral composition[3]. It has abundant amino acids, including the eight essential ones for the human body.A variety of bioactive substances such as polysaccharides, vitamins,and proteins, were detected and conf irmed to be benef icial to human health[4]. It was found that the active ingredients extracted from the whiteH. marmoreushad the properties of inhibiting leukemia and melanoma, lowering blood sugar, resisting inflammation, reducing oxidation, anti-aging, and others[5-8].
TheH. marmoreusis one of the three major industrialized species,next toFlammulina velutipesandPleurotus eryngii, in China[9-10].However, the development of the edible mushroom industry is greatly limited by some diseases caused by molds, bacteria, and viruses[11-13].Molds are the main pathogens appearing in the mycelial growth phase of the mushroom cultivation process. For example,Paecilomyces penicillatuscauses white mold disease, which is one of the most serious diseases in theMorchella esculentacultivation process[14].Fungal diseases often occur in the cultivation ofGanoderma lucidum,most commonly caused byTrichoderma hengshanicum, resulting in the rot and wilting of the fruiting body[15].T. atroviridecould also infectLentinula edodeswith serious effects[16].Cladobotryum mycophilumcauses cobweb disease inAgaricus bisporus[17].Without exception, these diseases cause yield loss and decrease the quality of mushrooms. Mold infection is also one of the main causes of yield reduction during the factory cultivation of the whiteH. marmoreus. In addition to the common pathogenPenicillium, there areFusarium,Aspergillus fumigatus,A. niger,A. flavus, and so on.
At present, most of the research on edible fungi diseases is focused on the identification of pathogenic factors, the classification and identification of pathogenic strains from the morphological and molecular levels, and exploring the pathogenic mechanisms of these molds. A study has shown thatC. protrusum, which can cause cobweb disease in cultivated edible fungi, has the potential to produce bioactive secondary metabolites and stress-response-related proteins. These products are of great significance for adaptation to harsh environments[18]. Another study provided insight into the molecular mechanism ofPenicilliumcausingM. esculentathe white mold disease. The genome ofPenicilliumcontains many gene clusters involved in the production of secondary metabolites with antifungal, antimicrobial or insecticidal activities. It was proved that thePenicilliumdid secrete some soluble compounds, which inhibited the growth of theM. esculentamycelium. These substances may have been responsible for the white mold disease[19]. Instead of destroying the cells of the mushrooms, these molds act as biological stress factors, inhibiting their growth of them. And how edible fungi respond to this stress exposure became a new idea in the research of mushroom diseases. Some chemicals produced by pathogens might change the quality and composition of the mushroom culture medium and cause mushroom mycosis by inhibiting the growth of mushrooms[20]. A study analyzed by gas chromatography-mass spectrometry (GC/MS) combined with liquid chromatography-mass spectrometry (LC/MS) showed that extracellular metabolites ofP. ostreatusmycelium affected by high temperature could promote the growth ofT. asperellum. And this fungus grew rapidly by competing for exogenous nutrients to occupy the ecological niche. Thus, the growth ofP. ostreatuswas inhibited[21]. The volatile compounds produced byH. marmoreus(TUFC 11906) have certain antifungal activity, indicating the existence of stress responses against the infection of pathogens in this mushroom[22]. At present, how edible fungi respond to the biotic stress of pathogens needs to be further studied, and the interaction mechanism between them needs to be improved.
Fungal cellular stress responses are related to the fundamental processing and stability of proteins and DNA, and developments in the field of proteomic analysis have provided insights to systematically understand the complex developmental events that occur in fungi at the molecular level in response to cellular stress exposure. The two-dimensional liquid chromatography-tandem mass spectrometry (LC-MS/MS) is one of the LC-based proteomic research techniques. It has been used in many studies to analyze proteomic changes during the complex development of edible fungi and has become a more suitable tool for this kind of research[23]. This technique was used to analyze the proteomes covering the three key stages ofOphiocordyceps sinensisfruiting body development and the underlying biological causes ofO. sinensisfruiting body development and plateau adaptation, thus providing information for this valuable fungal industry[24]. To gain insight into the mycelial growth and fruiting body formation events ofFlaintnulina velutipesand promote the identification of potential stage-specific biomarker candidates,some studies have investigated the proteomic response of its mycelia to cold stress. To provide valuable evidence for better understanding the molecular mechanism of mycelium resistance to cold stress and fruiting body formation of fungi[25]. To identify the changes in protein expression during the ripening process of fruiting bodies after harvest,LC-MS/MS for relative and absolute quantification was used in combination with bioinformatics analysis to understand the ripening process ofA. bisporus.It may help develop breeding programs to identify strains with extended shelf life[26].
Weighted correlation network analysis (WGCNA), an omics analysis method, can be applied to analyze highly multivariate and complex data, where highly correlated clusters are grouped into the same module. WGCNA can also be used to construct gene (protein)interaction networks, where each node represents a gene (protein) and the line connecting two nodes indicates the co-expression correlation between the two genes (proteins). The most mutual links shown in the network are considered to be the central genes (central proteins). This method has been more and more applied to the research of multiple infectious diseases. In the analysis of transcriptome data from young fruiting body samples ofF. velatumwith a blight disease, WGCNA was used to reveal the important gene modules with high correlation with disease traits and establish the networks of gene interactions[1].In the exploration of the molecular mechanism of defense reaction mediated by the rice blast resistance genes, two modules related to photosynthesis and important agronomic traits with similar expression patterns (SEP) were selected with the aid of WGCNA analysis.The important function of the modules in the resistance reaction was recognized by module gene function analysis[27]. WGCNA was also used in a study to reveal the molecular mechanism of the early responses ofEucalyptus grandisduring rust infection. The rustresistant and rust-prone genes were divided into several different modules, and it was revealed the active roles of metabolites and related enzymes in rust resistance caused by photosynthesis and oxidation-dependent reactions at different times by the functional analysis of the genes[28].
There is no proteomic analysis has been applied to study the biotic stress response mechanism of the whiteH. marmoreuscaused by fungal infections till this research. This study aimed to evaluate the overall change of protein expression patterns of the whiteH. marmoreusafter the mold infection through the label-free LC-MS/MS technology. Differentially expressed protein (DEPs) were selected and their functions were understood. They were most likely involved in stress response processes. WGCNA was used to screen out the protein modules that were highly correlated with disease traits, and the networks of the proteins interactions were established according to the relationship between proteins. The results pointed out the breeding direction of the whiteH. marmoreusto the mold infection disease resistance.
2. Materials and methods
2.1 Disease symptoms, pathogen purification, and identification
Mold contamination is a very common phenomenon in the whiteH. marmoreusindustry. The whiteH. marmoreusHW-7 strain (HW-7)taken from the Gujian Biology Co., Ltd. (Dongying, Shandong province) was used for research. It was often contaminated by a blue-green mold in the mycelium growth stage leading to serious competitive growth inhibition between the HW-7 and this mold. The HW-7 was more inhibited. And the mold contamination that occurred earlier, the more severely inhibited the HW-7. If the contamination occurred at the early stage of HW-7 mycelium growth, the large visible mold plaques could appear and the growth of HW-7 almost stopped resulting in the final failure to produce fruiting bodies. If at the mature stage, the mold could be presented as small visible plaques. The growth and development of HW-7 became slow with no normal mushrooms produced eventually.
The molds were identified by morphological and molecular biological methods. We took samples from different contaminated areas and different batches randomly. A small amount of these samples were dipped into the lactic acid carbolic acid cotton blue dyeing solution respectively to observe its microscopic appearance under a light microscope with a 40 × objective and 10 × eyepiece. It helped to roughly determine which genus it belonged to.
A small amount of mature mycelium was taken into a 5% Tween 80 aqueous solution, and impurities and mycelia were removed by filtration to obtain spores suspension. And it was diluted to the final concentration of 103spores/mL. Remove 100 μL from the diluted spores suspension and spread on the potato dextrose agar (PDA)(BD Difco). After the incubation in a constant temperature incubator at 25 °C for 48 h, several independent colonies appeared and then were transferred to the new PDA medium separately. After about a week, spores were collected again for single spore separation. This process was repeated several times until a pure culture of the mold was obtained. Subsequently, the colony morphological characteristics of mold were observed in terms of morphology, diameter, altitude,edge, surface appearance, density, concentration, and color.
The mold was identified according to its Internal Transcribed Spacer (ITS) sequence. The DNA of the mold was extracted with the aid of the Fungal Genomic DNA Extraction Kit (Solarbio Co., Ltd.). And it was used as a template to amplify the ITS1-5.8S gene-ITS2 region of fungal ribosomal RNA. The universal primers, ITS1: 5’-TCCGTAGGTGAACCTGCGG-3’ and ITS2:5’-TCCTCCGCTTATTGATATGC-3’, were used for the polymerase chain reaction (PCR)[29]. PCR reaction system: 3 μL DNA, 3 μL primer ITS1, 3 μL primer ITS4, 25 μL 2 × T5 Direct PCR highfidelity DNase (Tsingke Biotechnology Co., Ltd., Beijing, China),16 μL sterile water. Reaction conditions: 98 °C for 3 min; 98 °C for 10 s, 54 °C for 10 s, 72 °C for 15 s, 35 cycles; 72 °C for 2 min. The primer synthesis and PCR product sequencing were completed by Sangon Biotech Co., Ltd. (Shanghai, China). The sequencing data were uploaded to GenBank with accession numbers MK346198 and MK346199, respectively. The Blast search tool (https://www.ncbi.nlm.nih.gov/) was used to find the homologous sequences of the mold ITS gene. All the sequences of high similarity were put together to construct phylogenetic trees by using MEGA7 software and the Neighbor-Joining method[30].
2.2 Simulation of the infection process of the mold to the HW-7 mycelium in the laboratory
Because the mold mycelium, the HW-7 mycelium, and the medium are difficult to separate in the factory-produced samples, we designed experiments to simulate the infection process of the mold to the HW-7 mycelium. The pure cultures of the HW-7 and the mold were transferred to a PDA medium and cultured at 25 °C for 1 week.Twelve mycelium blocks of the same size and similar growth state were inoculated into the new PDA medium plates. Three of them were assigned to the control group without any treatment. Another three were placed in Group A, and the mold spores were spread around HW-7 at the same time. Another three of them were placed in Group B.When the colony diameter of the HW-7 was about 3 cm after 5 days of growth, the mold spores were inoculated around the colonies.The remaining three were attributed to Group C, and the mold spores were inoculated in the same way until the HW-7 had grown for 8 days and the colony diameter was about 6 cm. All the groups were placed in the incubator at 25 °C. After the HW-7 had grown for 15 days,the final growth status was recorded and the samples to be tested were obtained.
2.3 Obtain the samples to be tested
All the mycelium of the untreated HW-7 was scraped for the control. The outer edge of the HW-7 colony at the time of the mold spores inoculation was taken as the boundary. All the inward mycelium was scraped for the internal sample, and the outward new HW-7 mycelium was the outer sample.
2.4 Proteome sequencing
All the methods for protein extraction from the samples, peptide digestion, peptides, and proteins identification, and proteome detection performed by label-free LC-MS/MS quantitative proteomics analysis technology (Benagen Co., Ltd., Wuhan, China) refer to the previous work content[2].
2.5 Proteomic data analysis
2.5.1 Data standardization and screening of DEPs
Raw data analyzed by Proteome Discovery software suite version 2.0 (Thermo Fisher Scientific, San Jose, CA, USA) was normalized as follows. Firstly, missing values were supplemented (four sets of data, data with only one set was deleted, and missing values in data with two or three sets were supplemented, which is the k proximity method). Then, median standardization was executed on intensity data. Protein identification was supported by at least two unique peptides with a false discovery rate lower than 0.05. The clean reads were located in theH. marmoreusproteome database (proteome ID UP000076154) from UniProt. EdgeR was used to screen DEPs with|log2(fold change)| > 1 and false discovery rate (FDR) < 0.01.
2.5.2 Analysis of protein co-expression
The WGCNA was used to construct the protein modules, and evaluate protein connectivity. Eigenvalues were calculated for each module and used to test for correlations with the traits of each group of samples. Specific parameters were set as follows: outlier detection threshold was –2.5 and all the outliers from the expressed proteins were removed. The soft power was 18, which was an empirical value.The correlation algorithm is Bicor. The network type was signed. The specified maximum power to check was 30. The minimum module size for module detection was 25. TheR2cut for defining a scale-free network was 0.83. TheR2did not reach 0.85, which may be caused by the large variation between the samples. This variation may be caused by different batches, sample heterogeneity, or excessive influence of experimental conditions on expression. The presence of abnormal samples could be checked by sample clustering and heat map analysis.
Each group of samples corresponds to a trait, and groups with similar traits will be clustered together. The groups that came together were looked at as a whole. Multiple protein modules with the highest relationship with the corresponding traits could be screened out. And DEPs in the highest correlated modules might play more critical regulatory roles, which might be involved in a variety of important metabolic or signal transduction processes. So the co-expression network of these DEPs would be constructed by Cytoscape[31]. In the network, the DEPs with significantly higher connectivity were hub proteins and the others were hub-surrounding proteins. The hub-surrounding co-expressed with the hub proteins with the weight of edges between them being greater than 0.1.
2.5.3 Bio-information analysis
TBtools was used to construct heatmaps and enrichment analysis.UpSetR is an open-source R package to show intersections of sets,their size, and other properties by employing a scalable matrix-based visualization, which is available at https://github.com/hms-dbmi/UpSetR/ (accessed on 1 October 2021). String (https://cn.string-db.org/cgi/), a search tool for the retrieval of interacting proteins, was used to accurately discover and annotate all functionally interacting proteins and assist in the construction of the co-expression networks.TheH. marmoreusproteome was annotated and functionally enriched using the UniProt. The DEPs was annotated and functionally enriched using the Gene Ontology (GO) tool (http://geneontology.org; accessed on 1 October 2022) according to 3 categories of cellular component(CC), molecular function (MF) and biological process (BP). We performed GO analysis by using the R-ggCyto tool. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of candidate proteins were retrieved by blasting against the KEGG database addressed to https://www.genome.jp/kegg/pathway.html (accessed on 1 October 2022) and were mapped to pathways.
2.5.4 The proteomic data were validated by real-time quantitative PCR (RT-qPCR)
The untreated HW-7 and the outer sample were used for RNA preparation. Total RNA and cDNA were prepared according to the previous description[32]. The threshold cycle (Ct) values were normalized to the expression level of the 18S rRNA gene[23].RT-qPCR reactions were performed in a volume of 15 μL on a LightCycler®96 real-time PCR System (F. Hoffmann-La Roche Ltd.,Basel, Switzerland) with ChamQ Universal SYBR qPCR Master Mix (Sparkjade, Shandong, China) according to the manufacturer’s instruction. Expression levels were calculated according to the 2–ΔΔCtmethod.
2.6 The effect of the mold products on the HW-7 mycelium growth
2.6.1 Water-soluble components
Two mold water-soluble product samples, L1 and L2, were used for this experiment. The mold spores were inoculated into sabouraud dextrose broth (SDB) (BD Difco) and incubated at 25 °C, 200 r/min for 14 days. The fermentation broth was taken as L1. Scraped off all the mold mycelium growing on the PDA after the incubation at 25 °C for 7 days. Freeze-dried the mycelium and ground it into powder, then extracted the water-soluble products with 5 mL sterile water for 1 day.The filtered liquid was L2. PDA medium plate with four pores was made in advance with the help of Oxford cups. The distance between adjacent pores was equal. In the center of the four pores, the HW-7 was inoculated. The distances between each pore and the HW-7 were equal and about 2–2.5 cm. 200 μL of L1, L2, SDB, and sterile water were removed and added to the pores separately. The SDB and sterile water were controls. Three replicates were made and incubated at 25 °C. When the HW-7 mycelium touched the outer edge of the Oxford cup, the experiment was stopped and the results were recorded.
2.6.2 Gases
The PDA medium plate bearing HW-7 colony of about 3 cm in diameter and the untreated PDA plate or covered with mold paired up together and acted as a lid for each other. The pair with untreated PDA plate served as a control. Three replicates were made for each group and sealed, and incubated at 25 °C. The experiment would be stopped and the results would be recorded until some of the HW-7 mycelia touched the inner edge of the plate.
2.7 Detection of the gases by GC-MS
The gases from the sealed space of the paired-up plates were extracted with a solid-phase microextraction needle (75 μm CAR/PDMS/DVB extraction fiber head, SUPELCO, USA) for 12 h,and desorbed at the injection port of the GC-MS for 5 min before detection. The detection instrument was Agilent 7980A GC-MS:5975C with a PAL System three-in-one sampler (CTC, Switzerland).The parameters were set: the energy of the EI ionization source was 70 eV; ion source temperature was 230 °C; quadrupole rod temperature was 150 °C; connector temperature was 250 °C; scan range was 30–400m/z. Operation conditions: the chromatographic column was HP-INNOWAX capillary column (30 cm × 0.25 mm ×0.25 μm); the carrier gas was He, the flow rate was 1 mL/min, and the separation ratio was 5:1; the injection temperature was 250 °C.The heating procedure: start at 40 °C and keep it for 5 min; the temperature raised to 250 °C at a speed of 8 °C/min; kept it for 5 min.
3. Results
3.1 The most common pathogen was purified and identified as a species of Penicillium.
In contrast to the uncontaminated samples (Fig. 1A), which eventually produced normal mushrooms, most of the contaminated samples showed the same symptoms with the large areas of mold plaque (Fig. 1B). The pure cultures of the molds shared the same morphology. They were all large, grayish-green, fluffy colonies(Fig. 1C). It was shown that all the molds belonged toPenicilliumthrough the observation of the microscope (Fig. 1D). Their ITS sequences were determined to be identical, so they were the same species and temporarily namedPenicilliumsp. No1.

Fig. 1 The morphology changes of the HW-7 after mold infection and the pathogen was purified and identified as Penicillium. (A) The uncontaminated HW-7; (B) The contaminated HW-7 by mold; (C) The purified pathogen; (D) The pathogen was Penicillium; (E) The phylogenetic tree showed a relationship to other Penicillium.
The phylogenetic tree of the ITS sequences ofPenicilliumsp.No1 and its homologs showed their kinship (Fig. 1E and Table S1).The standard strains of the same species will cluster on a branch,and different species are located on different branches at a certain distance.Penicilliumsp. No1 was clustered withP. citrinumNRRL 1841 andP. hetheringtoniiCBS 122392 to a clade, with a bootstrap value of 50%. So this attribution to this branch was credible. The similarity of ITS sequences betweenPenicilliumsp. No1 and these two strains was 97.54%, which could be considered as the same species, but different strains.
3.2 Simulation of the infection process of Penicillium sp. No1 to the HW-7 in the lab
The HW-7 mycelium of Group A stopped growing (Fig. 2A), so this sample could not be obtained;Penicilliumsp. No1 mycelia of Group B occupied the blank area rapidly, and the growth of HW-7 mycelia grew very slowly, the mycelium inside the boundary was named SmI, and the mycelium outside the boundary was named SmO(Figs. 2B–B4).Penicilliumsp. No1 of Group C grew quickly to fill the blank area, and the HW-7 mycelia growth slowed down. But the HW-7 continued to spread beyondPenicilliumsp. No1 (Figs. 2C–C4).The mycelium within the boundary was named LaI, and the mycelium outside the boundary was named LaO. The earlier the infection was,the more serious inhibition on the HW-7 mycelium growth. The untreated HW-7 was named CK, which could grow into white hyphae with uniform texture and quickly cover the whole plate (Fig. 2D).The results of the simulation experiment were consistent with the actual phenomenon in the factory production process, so the samples obtained in the lab could be used for subsequent research.

Fig. 2 Different inhibitory effects were formed when Penicillium sp. No1 infected the HW-7 at different times. (A) The growth of the HW-7 was stopped when Penicillium sp. No1 and the HW-7 were inoculated simultaneously. (B) The Penicillium sp. No1 was inoculated in the blank space with the HW-7 colony diameter of about 3 cm after 5 days of growth. (B1, B3) The experimental Group B continued to grow for 10 days. The inner mycelium was SmI and the outer was SmO.(B2, B4) The back of the B1 and B3 respectively. (C) The Penicillium sp. No1 was inoculated with the HW-7 colony diameter of about 6 cm after 8 days of growth.(C1, C3) The experimental Group C continued to grow for 7 days. The inner mycelium was LaI and the outer was LaO. (C2, C4) The back of the C1 and C3 respectively. (D) The control named CK was the unprocessed HW-7.
3.3 Proteins sequencing and samples grouping
Total proteins from the control group (CK) and case groups(SmI, SmO, LaI, and LaO) were extracted and sequenced by labelfree LC-MS/MS quantitative proteomics analysis technique. After the sum-normalization of the data, 2 654 total proteins could be used for analysis (Table S2). Pearson correlation coefficient analysis (PCCA, Fig. S1A) and principal component analysis (PCA,Fig. S1B) both showed that the correlation of samples within each group was high, indicating little difference among duplicate samples.And all the correlations between groups were low, indicating significant differences between groups. These results indicated that the data were valid and could be used for further analysis. The phylogenetic tree showed that duplicate samples in the same group clustered on one terminal branch, and different groups were located on different branches at a certain distance. CK and LaI were clustered on one clade. The SmI, SmO, and LaO were clustered on another clade, and the SmO and LaO were more closely related (Fig. S1C).The groups of samples clustered in the same clade could be analyzed as a whole. The LaI was significantly different from the others, which was closer to the CK.
3.4 Screening of the DEPs
Compared with the CK group, the proteins with |log2(fold change)| ≥ 1 were selected as DEPs. Fold change is equal to the protein amount of the case groups or CK group. There were 141 DEPs screened out in the SmI, including 80 up-regulated and 61 down-regulated ones.There were 258 DEPs in the SmO, including 145 up-regulated and 113 down-regulated, 39 DEPs in the LaI, containing 24 up-regulated and 15 down-regulated, and 219 DEPs were identified in the LaO with 138 up-regulated and 81 down-regulated proteins included (Fig. S2A).Some DEPs were repeated in the four case groups. The total number of up-regulated DEPs was 256 (Fig. S2B), and the number of downregulated was 150 (Fig. S2C and Table S3).
The protein and mRNA abundances of the same gene were more likely to be consistent. To confirm the protein differences at the transcript level, the abundance of mRNA corresponding to 10 key upregulated DEPs was detected. Primers for RT-qPCR analysis were listed (Table S4). The results showed that all these mRNA expression levels were significantly up-regulated in the LaO group compared with those in the CK group, which exhibited a consistent correlation between the mRNA and protein expression abundance changes(Fig. S3 and Table S5). The results illustrated that the proteomic data was valid and the definition of the DEPs was accurate.
3.5 The analysis for protein co-expression network
WGCNA clustered highly correlated proteins to obtain 21 different modules, which were distinguished by different colors, as shown in the correlation plot of the module protein and trait (each case group corresponded to a trait). The distribution of DEPs was shown by the heat map (Fig. 3A and Table S6). WGCNA calculated the correlation between the modules and the combination of traits (SmI, SmO, and LaO). It was found that the MEgreenyellow (trait correction 0.859,P= 0.000 042), the MEgreen (trait correction 0.769,P= 0.000 808),and the MEsalmon (trait correction 0.690,P= 0.004 443)modules were highly associated with these three cases groups. These three modules were also highly related to the combined traits of the SmO and the LaO (Fig. 3B and Table S7). It was also found that the three modules were correlated with each other (Fig. S4). They were most likely to be highly related to the growth conditions formation or the biological stress response for the case groups. There were 23 DEPs in the MEgreenyellow, 33 DEPs in the MEgreen, and 15 DEPs in the MEsalmon. Interestingly, the DEPs were all up-regulated ones,so they were more likely to be responsible for the stress response.And the hub DEPs might play a central role in this response process.

Fig. 3 WGCNA clustered functionally highly correlated proteins into the same module and the modules highly related to the traits of the case groups were screened out. (A) The correlation plot of module protein and trait. It showed all the expressed proteins grouped into 21 different modules together with assigned colors. The heatmap showed the spread of the DEPs in different modules in each group. (B) The heatmap of module and trait. With the same color legend, three of the modules, MEgreenyellow, MEgreen, and MEsalmon labeled with red stars, were identified as highly correlated with the traits of the SmI, LaO, and SmO groups. The upper number is the Person correlation value and the number below is the P-value, of the module and trait.
The String database was used to annotate the functions of the proteins in the three modules and retrieved the protein interaction relationships. The minimum required interaction score of the confidence was set to be 0.1, and the protein-protein interaction networks were constructed. Thirteen DEPs in the MEgreenyellow module were connected into a network, and 7 of them were selected as hub DEPs with high connectivity values. The hub DEPs were the glucose-6-phosphate 1-dehydrogenase (gsdA, ID: A0A369JND0),the phosphate dehydrogenase (aldA_0, ID: A0A369KEH5), the putative NAD(P)H-dependent D-xylose reductase xyl1 (xyl1,ID: A0A369K2V1), the disulfide-bond oxidoreductase YfcG(yfcG_2, ID: A0A369JS89), the ornithine aminotransferase (car-2,ID: A0A369JFW8), the acetyl-coenzyme A synthetase (ACS-1,ID: A0A369JA05), and the putative glutathione S-transferase(GSTX4, ID: A0A369K1T7). The rest were hub-surrounded DEPs(Fig. 4A and Table S8). Twenty-three DEPs in the MEgreen module were connected to another network. Seven of them were hub DEPs, including the ATP-dependent RNA helicase ded1 (drh-9,ID: A0A369JUR0), the UV excision repair protein rhp23 (rhp23,ID: A0A369J6G9), the heat shock protein (ID: A0A369K2K3),the NADP-specific glutamate dehydrogenase (GDHA, ID:A0A369JDW9), the histone H3.2 (HHT2_5, ID: A0A369JT59), the histone H4 (H4.1_0, ID: A0A369J1D5), and the uncharacterized protein (ID: A0A369JP02) (Fig. 4B and Table S8). Eight DEPs in the MEsalmon module were connected into another network, and 5 hub DEPs were the NAD-specific glutamate dehydrogenase (gdh-1, ID:A0A369J9X4), the glutamine synthetase (GLN1, ID: A0A369JN44),the glutathione reductase (gtr-1, ID: A0A369JWV1), the 5-aminovalerate aminotransferase DavT (davT, ID: A0A369JZX8),and the putative aspartate-semialdehyde dehydrogenase (ID:A0A369JF26) (Fig. 4C and Table S8).

Fig. 4 The co-expression network diagrams of the DEPs from the (A) MEgreenyellow, (B) MEsalmon, and (C) MEgreen modules. The circle nodes represent proteins and the straight lines represent edges. Node size stands for the connectivity value of proteins and the width of a straight line stands for the weight value.Orange nodes represent hub DEPs with a certain function, the blue node represents a hub DEP annotated as a hypothetical protein, and the green nodes represent hub-surrounded DEPs.
3.6 Functional annotation of DEPs
GO analysis was used to classify DEPs to show the changes in protein function for the case groups. The annotated portion of the total up-regulated DEPs was mainly assigned to 26 GO terms(P< 0.01). Nineteen GO terms were classified into the MF category,mainly including the terms of oxidoreductase activity (count: 17,ratio: 6.64%), heme binding (14, 5.47%), metal ion binding (14,5.47%), iron ion binding (13, 5.08%), flavin adenine dinucleotide binding (12, 4.69%), oxidoreductase activity acting on paired donors with incorporation or reduction of molecular oxygen (12, 4.69%)and monooxygenase activity (8, 3.13%). There were 4 terms in the CC category, and the most representative ones were cytoplasm (10,3.91%) and nucleosome (4, 1.56%). Three terms were included in the BP category, of which proteasome-mediated ubiquitin-dependent protein catabolic process (3, 1.17%) was the main one. These results showed that the dominant molecular functions of the proteins were related to oxidoreductase activity. These proteins might be related to the stress response of the HW-7, such as the response to its growth inhibitor, cellular detoxification, and cellular oxidative stress. The DEPs of the MEgreenyellow module were mainly concentrated in the terms (P< 0.01) of oxidoreductase activity (MF, GO: 0016491),NADP binding (MF, GO: 0050661), and pyridoxal phosphate binding(MF, GO: 0030170), which might be involved in REDOX processes.The DEPs of the MEgreen module were mainly classified into the terms (P< 0.01) of proteasome-mediated ubiquitin-dependent protein catabolic process (BP, GO: 0043161), protein heterodimerization activity (MF, GO: 0046982), transaminase activity (MF, GO:0008483), pyridoxal phosphate binding (MF, GO: 0030170), NADP binding (MF, GO: 0050661), and methionine biosynthetic process(BP, GO: 0009086), which might be related to the process of amino acids and protein metabolism. The DEPs of the MEsalmon module,mainly including NADP binding (MF, GO: 0050661), transaminase activity (MF, GO: 0008483), and pyridoxal phosphate binding (MF,GO: 0030170) terms (P< 0.01), might be associated with amino acids metabolism (Fig. 5A and Table S9).
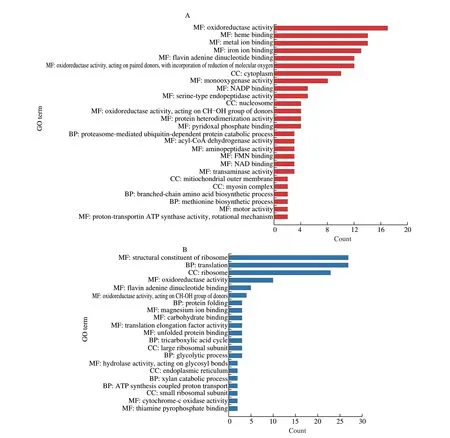
Fig. 5 Enriched GO terms of the most DEPs were presented using bar plots. (A) GO for the DEPs with up-regulated expression. (B) GO for the down-regulated DEPs. The ordinate indicates multiple terms in different categories. MF, molecular function category; BP, biological processes category; CC, cellular component category. The abscissa indicates the count of DEPs in each term.
Total down-regulated DEPs were primarily assigned to 21 GO terms (P< 0.01), 11 of them were in the MF category, 6 in the BP category, and 4 in the CC category. The terms in the MF category were dominantly structural constituents of ribosome (27, 18.00%)and oxidoreductase activity (10, 6.67%). In the CC category, the main GO terms were involved in the ribosome (23, 15.33%) and the large ribosomal subunits (3, 2.00%). In the BP category, DEPs were enriched in the basic metabolic processes of life, such as translation(27, 18.00%) and glycolytic process (3, 2.00%). These results showed that the number and function of ribosomes in the case groups were decreased, which was consistent with the growth retardation of HW-7. It was indicated that the ribosome acted as the terminal responder to biological stress. The weakening of the glycolytic process, which might involve the molecules with oxidoreductase activity, was also consistent with the growth retardation (Fig. 5B and Table S9).
3.7 KEGG pathway enrichment analysis for the DEPs
To obtain functional information on DEPs in metabolic pathways,KEGG pathway enrichment analysis was performed. Of the total up-regulated DEPs, 73 DEPs were labeled to 80 specific KEGG pathways (P< 0.05). It mainly involved the pathways of biosynthesis of amino acids; valine, leucine, and isoleucine degradation/biosynthesis arginine and proline metabolism/biosynthesis;phenylalanine, tyrosine and tryptophan biosynthesis/metabolism;lysine biosynthesis/degradation; alanine, aspartate, glutamate,cysteine, methionine, glycine, serine, threonine, histidine, beta-alanine and taurine metabolism; biosynthesis/metabolism of cofactors; carbon and carbohydrates metabolism fatty acid degradation/metabolism;oxidation and energy metabolism; protein processing/degradation;glutathione metabolism; nitrogen/amine/ammonia metabolism;nucleotide metabolism/DNA replication, repair, transport; RNA surveillance/degradation; propanoate metabolism; autophagy;endocytosis; cell division; sulfur metabolism; secondary metabolites;basal transcription factors; porphyrin metabolism; ABC transporters;mitogen-activated protein kinase (MAPK) signaling pathway;selenocompound metabolism. These enhanced metabolic pathways were used to indicate the response processes of the HW-7 to the stress caused by the mold (Fig. 6A and Table S10).
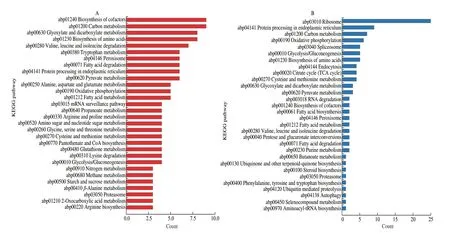
Fig. 6 KEGG pathway enrichment of the most DEPs was shown by bar plots. (A) The KEGG pathways involved by the DEPs with up-regulated expression. (B) The KEGG pathways involved by the down-regulated DEPs. The ordinate indicates multiple KEGG pathways. The abscissa indicates the count of DEPs in each pathway.
There were 38 down-regulated DEPs labeled to 41 specific KEGG pathways (P< 0.05). The main attenuated pathways were ribosome; protein processing in endoplasmic reticulum; spliceosome;carbon metabolism/glycolysis/gluconeogenesis; oxidation and energy metabolism; biosynthesis/metabolism of amino acids; fatty acid metabolis; endocytosis; proteolysis; purine metabolism; biosynthesis of cofactors; RNA degradation; aminoacyl-tRNA biosynthesis;selenocompound metabolism; autophagy; steroid biosynthesis;ubiquinone and other terpenoid-quinone biosynthesis; butanoate metabolism; porphyrin metabolism; nucleotide metabolism; protein export; MAPK signaling pathway; cell cycle; phosphatidylinositol signaling system . The attenuated pathways were mainly involved in the synthesis of cell components, which was related to the growth and development of the HW-7 mycelium. The attenuation of these pathways was consistent with the growth retardation (Fig. 6B and Table S10).
4. Discussion
The stress response of living cells was directly related to the expression, processing, and stability of proteins[33]. Developments in the field of comparative proteomic analysis over the past decade have provided insights into the systematic understanding of the developmental states that fungi exhibit at the molecular level in response to complex environments[34-35]. However, the proteome changes of edible fungi under biological stress have not been fully interpreted. In previous work, we studied the proteome of the whole process of growth and development of HW-7 by using LC-MS/MS technology and obtained a relatively comprehensive understanding of its genetic background and developmental mechanism[2]. This made the HW-7 a potential model system for studying the basic mechanism of cell stress response in edible fungi. In this study, the proteome changes of HW-7 mycelium under the stress caused byPenicilliumsp. No1 were studied to explore the pathogenic mechanism and stress response mechanism of edible fungi under the mold attack.Our data suggested that some interesting DEPs whose expression is up-regulated might play a key role in the stress response of HW-7,which was closely related to external factors that inhibited or retarded the HW-7 mycelium growth. However, the proteins with downregulated expression might be connected with the processes such as the synthesis of the cell components, cell proliferation, energy metabolism, etc., which was the internal reasons for the severe restriction of the HW-7 mycelium growth.
4.1 The exploration for the external reason of severe restriction of mycelia growth caused by the Penicillium sp. No1
Penicilliumsp. No1 grew faster and could occupy a favorable ecological niche, which was an important reason for the growth restriction of the HW-7 mycelium in the confrontation growth process of these two fungi. Fungi within a certain space could also produce some special metabolites to help them compete for limited nutrients and space[36-38]. The interaction between the HW-7 andPenicilliumsp. No1 did not cause tissue damage to each other, which indicated that they might produce some sensitization components that affect each other, thus showing such a competitive relationship. It has been shown thatPenicilliumcould produce some components to inhibit the growth of edible fungi[39]. However, when the watersoluble metabolites produced byPenicilliumsp. No1 were applied to the HW-7 mycelium, it was found that the growth of HW-7 was not inhibited (Fig. S5). This result indicated that the water-soluble products did not play a significant role in this inhibition process,and which specific components were responsible need to be further explored.
At the same time, we also explored the influence of gases produced byPenicilliumsp. No1 on the HW-7 mycelium growth.It was found that the growth of HW-7 could not be inhibited(Fig. 7A–A2) without the existence of thePenicilliumsp. No1, while the HW-7 was severely inhibited when thePenicilliumsp. No1 existed without the mycelium of the two fungi contact with each other(Fig. 7B–B2), and the inhibition rate reached 53% (Fig. 7C and Table S11).The gases produced byPenicilliumsp. No1 alone, the HW-7 alone,and these two fungi were detected with the aid of GC-MS. By comparing with the gases produced by the pure culture of the HW-7 andPenicilliumsp. No1 respectively, and the paired-up of these two fungi, some of the gas molecules produced byPenicilliumsp. No1 that had an inhibitory effect on the HW-7 were mainly ethylbenzene,p-xylene, 1,3-dimethyl-benzene, 1,3,5,7-cyclooctatetraene,1,2,3-trimethoxybenzene, 1,2,4-trimethoxybenzene, 1,2-benzenedicar boxylic acid, butyl 2-methylpropyl ester, dibutyl phthalate and so on (Fig. S6). The benzene compounds were chemical pollutants,and their oxidative metabolites of them were involved in oxidative damage causing cytotoxicity[40]. The results above suggested that these gases produced byPenicilliumsp. No1 might be another major factor leading to the growth restriction of the HW-7 mycelium.
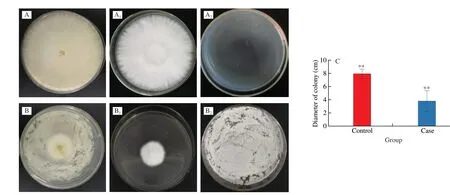
Fig. 7 The gases produced by Penicillium sp. No1 inhibited the HW-7 mycelium growth. (A) The HW-7 paired up with the blank medium. (A1) The HW-7 from A.(A2) The blank medium from A. (B) The HW-7 paired up with the Penicillium sp. No1. (B1) The HW-7 from B. (B2) The Penicillium sp. No1 from B. (C) The bar chart showed the diameters of the two groups of the paired-up samples. The diameter of the HW-7 from A regarded as the control group was 8.0 cm. And the diameter of the HW-7 from B viewed as the case group was 3.8 cm. **P < 0.01.
4.2 Detoxifying proteins directly transformed harmful substances and played a vital role in the stress response of the HW-7
Aryl-alcohol dehydrogenase (AAD) and aldehyde dehydrogenase(AD) had detoxification functions. In a variety of lignin hydrolyzing fungi, it was found that the cytochrome P450, aryl alcohol dehydrogenase, and aryl aldehyde dehydrogenase were all involved in the biotransformation of polychlorinated biphenyl (PCBs) into chlorobenzene acid, chlorobenzene aldehyde, and chlorobenzene alcohol, which acted as detoxification enzymes[41]. Interestingly, we found 5 aryl-alcohol dehydrogenases (A0A369J8X4, A0A369JQM8,A0A369J218, A0A369K712, and A0A369J2C4) among the significantly up-regulated proteins. They might be involved in the transformation of the benzene compounds in the cells. AAD has the function of reversely catalyzing alcohol dehydrogenation to form aldehydes (or ketones). In the white rot fungusBeauveria bassiana,the function of AAD had the activity of reducing aryl aldehydes and linear aldehydes with NADPH as a coenzyme[42]. The AAD in the yeastSaccharomyces cerevisiaeBY4741 had the activity on veratraldehyde, cinnamaldehyde and vanillin, so they could detoxify the aromatic aldehyde released during the degradation of lignin[43].The AAD might reverse the conversion of aldehydes into alcohols when the concentration of aldehydes was high in the cells. Similarly,the protein aldehyde dehydrogenase (A0A369KEH5) with an upregulated expression appeared in the arginine, proline, pyruvate,tryptophan, beta-alanine, and histidine metabolism (abp00330;abp00620; abp00380; abp00410; abp00340), the biosynthesis of cofactors (abp01240), the valine, leucine, isoleucine, and lysine degradation (abp00280; abp00310), the fatty acid degradation(abp00071), the glycolysis/gluconeogenesis (abp00010), the pantothenate and CoA biosynthesis (abp00770), the glycerolipid metabolism (abp00561), and the ascorbate and aldarate metabolism(abp00053) pathways. AD has been verified to be a key enzyme in detoxification, which might protect the integrity of cell membranes by scavenging reactive aldehydes, fatty acid-free radicals, and other alcohol derivatives. Therefore, it played an important role in the REDOX reaction and the oxidative stress responsein vivo[44-46]. AAD and AD might work in concert and participate in the detoxification process of the benzene and its transformed aldehyde intermediates.
The aldo-keto reductase yakc [NADP+] (AKR, A0A369JQN1),the 4-O-methyltransferase (A0A369K1V0), and the benzoate 4-monooxygenase (A0A369K8D3) might participate in the transformation process of aldehydes, ketones, and phenols. The AKR was a superfamily of NAD(P)H-linked REDOX enzymes that help eliminate cellular stress, such as reactive oxygen species(ROS) and lipid peroxides, produced by some drugs. The AKR converted aldehydes and ketones produced during the heterogeneous biological metabolisms to reduce them to their respective primary and secondary alcohols[47]. 4-O-Methyltransferase detoxified by converting free hydroxyl phenol into methylated phenol[48]. Benzoate 4-monooxygenase was a highly conserved protease in fungi, which was involved in relieving the toxicity produced by phenols[49].
4.3 The key role of glutathione metabolism in stress response
The glutathione metabolism played an important role in oxidative stress response, and there was a group of DEPs with up-regulated expression acting on the production of reduced glutathione (GSH) to reduce oxidative damage caused by various toxic substances. It was found that the hub DEPs in the MEgreenyellow module played a key role in glutathione metabolism.
The cells exposed to 1,4-benzoquinone (BQ) could significantly increase the expression of glucose-6-phosphate 1-dehydrogenase(G6PD), which was the first and rate-limiting enzyme of the pentose phosphate pathway[50]. G6PD could improve the ratio of NADPH/NADP and GSH/oxidized glutathione (GSSG). GSH has an antioxidant effect, and the sulfhydryl of cysteine on GSH was the active site, which was easy to combine with some drugs and toxins. It could also reduce the level of the relative ROS in cells and decrease the oxidative damage of DNA. Therefore, G6PD exhibited a strong detoxification ability by increasing the amount of GSH[51-52]. There was a G6PD (A0A369JND0) involved in carbon metabolism (abp01200), glutathione metabolism (abp00480), and pentose phosphate (abp00030) pathways. It was a hub DEPs of the MEgreenyellow module. The up-regulated expression of this enzyme was beneficial to increase the content of GSH, which could increase the antioxidant stress ability of the HW-7. In addition to G6PD(A0A369JND0) (1.1.1.49, 1.1.1363), There were also 2 hub DEPs in the MEgreenyellow module, the putative glutathione S-transferase(A0A369K1T7) (2.5.1.18) and the disulfide-bond oxidoreductase YfcG (A0A369JS89) (2.5.1.18), directly involved in glutathione metabolism (abp00480). In addition, the glutathione reductase(A0A369JWV1) (1.8.1.7), which belonged to the MEsalmon module,was also involved in glutathione metabolism (Fig. S7).
It was believed that there was a high correlation between proteins in the same module, so it was speculated that the other 4 hub DEPs in the MEgreenyellow module might be indirectly involved in the glutathione metabolism. The putative NAD(P)H-dependentD-xylose reductase xyl1 (A0A369K2V1), participating in pentose and glucuronate interconversions (abp00040), possibly involved in the pentose phosphate pathway. The ornithine aminotransferase car2(A0A369JFW8) was involved in the arginine and proline metabolism pathway (abp00330). In the process of proline metabolism, NADP+was produced, which was an essential component of pentose phosphate synthesis of NADPH. Therefore, the proline metabolism was conducive to the amount increase of GSH[53]. AD (A0A369KEH5)(1.2.1.3) and acetyl-coenzyme A synthetase (A0A369JA05) (6.2.1.1)participated in pyruvate metabolism pathway (abp00620) (Fig. S8).The increase of the pyruvate content could enhance the citric acid(TCA) cycle, and the increase of citric acid could inhibit the activity of phosphofructokinase, to the pyruvate entered the pentose phosphate bypass and the NADPH was produced, thereby indirectly increasing glutathione (GSH) content.
In most bacteria and plants, direct biosynthesis of cysteine from sulfides viaO-acetylserine (OAS) was essential for the production of sulfur amino acids from inorganic sulfur[54]. Thede novocysteine biosynthesis pathway inLeishmaniawas comprised of serineO-acetyltransferase (SOAT) and cysteine synthase (CS) enzymes which sequentially mediate two consecutive steps of cysteine biosynthesis with undebated roles in protection from oxidative damage and drug resistance[55]. There were a pair of DEPs with upregulated expression including SOAT (A0A369JCR2) and highly oxygenated organic molecules (HOM)-CS (A0A369K4Y3). The HOM-CS was involved in cysteine and methionine metabolism(abp00270), biosynthesis of amino acids (abp01230), sulfur metabolism (abp00920), and carbon metabolism (abp01200)pathways. The intracellular amounts of cysteine and glutathione were significantly reduced when the SOAT was absent, and the SOAT could regulate and reverse the disruption of cellular antioxidant balance due to H2O2-induced oxidative stress[56]. These two DEPs might regulate cysteine biosynthesis together.
It was worth noting that 2-oxoglutarate dehydrogenase (OGDH,A0A369JVG3) was a DEPs with significantly down-regulated expression. It participated in carbon metabolism (abp01200) and TCA cycle (abp00020) pathways. As a key REDOX sensor in mitochondria, OGDH was highly sensitive to peroxides such as 4-hydroxy-2-nonenyl, which could regulate the change of GSH and GSSG. The increase of OGDH promoted the increase of ROS leading to the consumption of GSH and the reduction of NADH. GSSG could reduce the OGDH activity and the corresponding ROS decreased[57-58].The increase of ROS during oxidative stress led to a wide range of secondary consequences, such as the damage of DNA or RNA,inflammatory damage, cell cycle arrest, and apoptosis, through the modification of cell signaling proteins[59]. Maintaining a stable and low concentration of ROS had positive effects on the growth, fruition of body development, the GSH/GSSG balance, and the carbon metabolism ofH. marmoreus[60]. The down-regulated expression of OGDH (A0A369JVG3) might be associated with reducing ROS production and avoiding GSH loss, thus lowering oxidative toxicity.
In conclusion, the HW-7 could maintain a high level of GSH through a multiple of DEPs related to the glutathione metabolism pathway to keep the strong antioxidant capacity of cells.
4.4 Histone modification, nucleotide excision repair, and removal of misfolded proteins might play an important role in the stress response
Histone modification played an important role in regulating transcription, recombination, DNA repair, DNA replication,chromatid cohesion, and chromosome separation. Histone methylation might play an important role in the complex fungal life[61]. It was shown that the fungal histone modification could regulate the growth and the infective ability of the plant pathogenic fungi[62]. There were two hub DEPs, the histone H4, partial (A0A369J1D5), and the histone H3.2 (A0A369JT59), in the MEgreen module. It was found that the expression of the putative protein lysine methyltransferase SET5 (A0A369JQ27) was also up-regulated. This kind of enzyme could target the methylation of histones or non-histones, which was associated with the stress response and genomic stability[63].
The ATP-dependent RNA helicase ded1 (ADRH, A0A369JUR0),the UV excision repair protein rhp23 (UV-ERP, A0A369J6G9), and the heat shock protein (HSP, A0A369K2K3) were also hub DEPs in the MEgreen module. They might be involved in correcting errors in DNA replication and transcription. ADRH has been shown to play a crucial role in DNA and RNA metabolism, such as replication,repair, recombination, transcription, translation, ribosomal biogenesis,and splicing. In plants, this ADRH might play an important role in plant growth under stable stress[64]. The UV-ERP was involved in the protein processing in the endoplasmic reticulum (abp04141)and the nucleotide excision repair (abp03420) pathway. The HSP (A0A369K2K3) took part in the RNA degradation pathway(abp03018). HSPs served as molecular chaperones to stabilize the function of various cellular components in fungi and played a vital role in cell survival, growth, virulence, and drug resistance[65].HSPs were a kind of protective protein for the response to a bad environment. They helped maintain normal physiological activity,prevent the interactions of the healthy proteins, the amino acid chains fold into the correct three-dimensional structure, remove the damaged and incorrectly folded peptides, escort proteins to the target sites, and so on[66].
There was a series of DEPs with up-regulated expression, the Lon protease, mitochondrial (A0A369K771), proteasome subunit beta type-6 (A0A369JU17), putative 26S protease subunit rpt4(A0A369JTI5), and 26S proteasome non-ATPase regulatory subunit 12 (A0A369K2S9), which might be involved in the removal of the ineffective proteins. Lon protease was found in the matrix of the mitochondria and degraded oxidized and misfolded proteins,thereby protecting cells from a variety of stresses, including oxidative stress[67-68]. Lon protease was also present in bacteria, which could remove the invalid proteins to cope with the toxic stress[69]. The A0A369JU17, A0A369JTI5, and A0A369K2S9 participated in the proteasome pathway (abp03050). The 26S protease was a kind of multi-catalytic protease complex, which could degrade the ubiquitinated proteins in eukaryotic cells. TheN-methylation of this kind of enzyme might be important for cell growth or stress tolerance in yeast[70]. And the putative protein lysine methyltransferase SET5(A0A369JQ27) might activate the function of the 26S protease.In addition, the putative nudix hydrolase P35G2.12 (NUDX,A0A369JJ34), participating in the purine metabolism (abp00230)and the riboflavin metabolism (abp00740) pathways, might play a role in the nucleic acids repair. The 8-oxy-(d) GTP produced by the ROS could be incorporated into DNA/RNA, resulting in replication and transcription errors. The NUDX could hydrolyze the 8-oxy-(d) GTP to reduce the level of oxidized nucleotides in cells[71]. The loss of the NUDX resulted in a decrease in glucose consumption, the decreased production of ethanol and lactic acid, and a reduced amount of ATP[72].
4.5 The key role of nitrogen metabolism in the nutrient deficiency stress response
There was a group of DEPs with up-regulated expression related to nitrogen metabolism, which might work together to resist the stress produced by nutrient deficiency. The hub DEPs in the MESalmon module were involved in nitrogen metabolism.
Multiple enzymes were involved in the conversion ofL-lysine toL-glutamate. InEscherichia coli, the addition of ethanol and hydrogen peroxide could enhance the production of 5-aminovalerate(5AVA) fromL-lysine[73].L-lysine decarboxylase (LdcC), putrescine transaminase (PatA), and C-aminobutyraldehyde dehydrogenase(PatD) participated in this transformation process[74]. The DEPs with up-regulated expression, including the putative decarboxylase tpcK(A0A369K823), the oxalate decarboxylase OxdC (A0A369K4T3),the ornithine aminotransferase car2 (A0A369JFW8), the putative aspartate-semialdehyde dehydrogenase (A0A369JF26) and other enzymes with the similar functions to the LdcC, PatA, and PatD,might play a role in the conversion ofL-lysine to 5AVA. The conversion of 5AVA into glutarate could be promoted by the 5AVA aminotransferase and glutarate semialdehyde dehydrogenase inPseudomonas putida[75-76]. It was found that two hub DEPs in the MESalmon module were highly related functionally and might be involved in the conversion of 5AVA into glutarate. The one was the 5-aminovalerate aminotransferase DavT (A0A369JZX8),participating in the pathways of the alanine, aspartate, and glutamate metabolism (abp00250) and the butanoate metabolism (abp00650).The other was the putative aspartate-semialdehyde dehydrogenase(ASADH, A0A369JF26). It was involved in the cysteine and methionine metabolism (abp00270), the glycine, serine, and threonine metabolism(abp00260), the 2-oxocarboxylic acid metabolism (abp01210), and the monobactam biosynthesis (abp00261) pathways. The conversion ofL-lysine toL-glutamate was accompanied by the production of glutarate.The glutarate was oxidized in the TCA cycle[77]. These enzymes worked together to increase the supply ofL-glutamate in cells.
There were another two hub DEPs in the MESalmon module, the NAD-specific glutamate dehydrogenase (NAD-GDH, A0A369J9X4)and the glutamine synthetase (GS, A0A369JN44). NAD-GDH and GS could convert glutamate into glutamine to provide a nitrogen source for nucleic acid biosynthesis for the cells. NADGDH participating in the glutamate metabolism (abp00250) and the nitrogen metabolism (abp00910) pathways, NAD-GDH could catalyze the formation of ammonium and 2-oxyglutaric acid from glutamate. The intracellular ammonium or metabolic intermediate could be used as a signal to induce the yeastSchizosaccharomyces pombeinto invasive growth[78]. GS was involved in the glyoxylate and dicarboxylate metabolism (abp00630), glutamate metabolism and the nitrogen metabolism pathways. GS could consume ATP and use the intracellular ammonium or glutamate to synthesize glutamine.It has been confirmed thatFusarium graminiferumresponded to nitrogen restriction signals of the external environment through the GS signaling pathway, and effectively activated the infection capacity, including mycotoxin production, vegetative growth,conidia, appressorium formation, cell wall integrity maintenance, and oxidative stress tolerance, ofF. graminiferum[79].
Penicilliumsp. No1 competed for nutrients, including nitrogen,resulting in restricted growth of the HW-7. The above enzymes could jointly promote the generation of the nitrogen sources that could be used directly by the HW-7 itself in response to the nitrogen restriction of the external environment.
4.6 The role of multiple different functional DEPs with upregulated expression in stress response
The succinyl-CoA:3-ketoacid coenzyme A transferase 1,mitochondrial (A0A369JUG2) participating in the valine, leucine,and isoleucine degradation (abp00280) and the butanoate metabolism(abp00650) pathways, played a role in oxidative stress by enhancing the activity of OxyR and catalase[80]. The 1,3-β-glucanosyltransferase gel4 (A0A369KA55) was a cell wall remodeling enzyme in fungi related to stress response, cell wall integrity, and virulence. There were multiple members contained in most fungal genomes[81-82]. These enzymes also played an important role in stress response.
4.7 When the growth of the HW-7 mycelium was restricted, various metabolic processes were adjusted to adapt to the changes
According to the multiple modes related to stress response,we could judge that the intracellular toxic molecules accumulated,more oxidative damages aggrandized, and damaged cells increased,nitrogen restriction formed in HW-7 when contaminated byPenicillium. All the changes led to the growth of HW-7 hyphae being slow. So the expression of some proteins related to mycelium growth and development would be adjusted.
4.7.1 The carbon metabolism and energy metabolism were reduced
In contrast to the DEPs with up-regulated expression, almost all dehydrogenases with down-regulated expression were related to carbon metabolism and energy productivity. This implied that these metabolisms were weakened in the growth-restricted HW-7. Two DEPs with down-regulated expression, the malate dehydrogenases(MDH; A0A369JS64 and A0A369JGA3), took part in the carbon metabolism (abp01200), the cysteine and methionine metabolism(abp00270), the TCA cycle (abp00020), the pyruvate metabolism(abp00620), and the glyoxylate and dicarboxylate metabolism(abp00630) pathways. MDH, a widely distributed enzyme catalyzing the conversion of oxaloacetate and malate, played key roles in many important metabolic pathways, such as the TCA cycle[83]. The downregulated protein OGDH (A0A369JVG3), also a highly regulated enzyme in the TCA cycle, could convert alpha-ketoglutaric acid(αKG) to succinyl-CoA with the NADH. And then produced ATP through oxidative phosphorylation. The dysregulation of OGDHc could impair cellular REDOX homeostasis, energy production, and amino acid metabolism[58,84]. The scyllo-inositol 2-dehydrogenase(NAD+) (SID; A0A369K8F2) with down-regulated expression,participating in the pentose and glucuronate interconversions(abp00040) pathway, was a key enzyme in hydroxyl oxidation[85]. The aldehyde-3-phosphate dehydrogenase (GAPDH, A0A369JU01) was a down-regulated protein that participated in the carbon metabolism(abp01200), the biosynthesis of amino acids (abp01230), and the glycolysis/gluconeogenesis (abp00010) pathways. The GAPDH was a highly conserved enzyme in the glycolysis pathway, which could catalyze the conversion of glyceraldehyde 3-phosphate to glycerol-1,3-bisphosphate[86]. TheD-arabinitol dehydrogenase (APDH,A0A369K9K8) was also a down-regulated protein, participating in the pentose and glucuronate interconversions (abp00040) pathway.The APDH could convertD-arabitol 1-phosphate (Ara-oll-P) to xylose-5-phosphate[87]. In addition, the cytochrome c oxidase (COX)was essential in mitochondrial ATP production. A0A369JL47 and A0A369JYI8 were both the mitochondrial COX subunits related to the oxidative phosphorylation (abp00190) pathway, and their expression was also reduced. The down-regulated expression of the above enzymes related to the carbon and energy metabolism was consistent with the growth restriction, which reflected the adaptation to the survival of the HW-7.
4.7.2 Effective protein production decreased.
There were 25 ribosomal proteins (A0A369JKV9, A0A369K9P8,A0A369JG46, A0A369JDR1, A0A369JQM2, A0A369JF09,A0A369KC73, A0A369K5V7, A0A369JBU0, A0A369K5L4,A0A369K666, A0A369KC14, A0A369JUZ4, A0A369KEW7,A0A369JN97, A0A369K9N8, A0A369KE53, A0A369K6L6,A0A369K0C0, A0A369JDX4, A0A369JM63, A0A369J7S7,A0A369J591, A0A369J7L9, A0A369JPJ9), correlated with ribosome(abp03010), with the down-regulated expression, which inevitably led to the decrease of the protein production. Simultaneously multiple down-regulated HSPs (A0A369JMQ9, A0A369KA26, A0A369JS72,A0A369JNN0, A0A369IY45, A0A369JVD2, A0A369JAL1,A0A369JGZ2, A0A369JUF6, A0A369KH52, A0A369JLF5)participated in the protein processing in endoplasmic reticulum(abp04141), the spliceosome (abp03040), the endocytosis (abp04144),and the RNA degradation (abp03018) pathways. The HSPs family had multiple members, and HSP90 played a role in fungal growth,cell survival, and pathogenicity. HSP70 and small HSPs were involved in fungal development, tolerance, and resistance to stressful conditions. HSP60 was a mitochondrial chaperone, and HSP40 regulated the activity of fungal ATPase[65]. The down-regulation of these HSPs weakened protein processing and maturation. Therefore the production of effective proteins for making up the cell structure was down-regulated, thus the cell division and extension, and the mycelia growth and development of the HW-7 were restricted.
The mechanism of the edible fungi response to biological stresses and the growth restriction adaptive mechanism discussed in this paper were summarized in Table 1. The mechanisms related response modes, the involved enzymes, the processes, and corresponding function or experimental phenomenon were presented.

Table 1 The biological stress response and growth restriction adaptation mechanisms to address the impact of Penicillium.
5. Conclusion
Penicilliumsp. No1 could produce harmful gases and occupy a favorable ecological niche to restrict whiteH. marmoreusHW-7 growth. By comparing the proteome of the HW-7 mycelium with different degrees of inhibition byPenicilliumsp. No1, it was found that there were significant differences. The DEPs with upregulated expression, mainly involved in the processes of non-toxic transformation of harmful gases, 4-O-methyltransferase, Aldo-keto reductase yakc [NADP+], and cell repair, played a key role in the stress response of HW-7. With the aid of WGCNA, three protein modules, the MEgreenyellow, MEgreen, and MEsalmon, highly related to the samples under stress were selected. The DEPs in MEgreenyellow mainly included the enzymes, which could participate in the glutathione metabolism and lower the ROS accumulation in cells, to reduce oxidative damage. The DEPs in the MEgreen mainly participated in the processes of histone modification, nucleotide excision repair, and the removal of the misfolded proteins to reduce the formation of wrong nucleic acids or proteins, which could eliminate the negative consequences caused by oxidative damage.The DEPs in the MESalmon were mainly involved in nitrogen metabolism to cope with the nitrogen restriction formed in the process of nutrient competition. The DEPs with down-regulated expression,mainly related to the processes of carbon and energy metabolism, the decrease in the effective protein production, might play a key role in the growth and development of the HW-7 mycelium. The reduction of these DEPs was an adaption to HW-7 with growth restriction. This study explored the reasons for the severe restriction ofH. marmoreusHW-7 caused byPenicilliumsp. No1, the stress response mechanism and the growth restriction adaptive mechanism of the HW-7 toPenicilliumwere also researched through comparative proteomics.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability statement
The raw data for the proteomic analysis reported in this paper was named “Proteomic changes in inhibited growth of the white Hypsizigus marmoreus caused by aPenicilliumstrain” and have been deposited in the OMIX, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (https://ngdc.cncb.ac.cn/omix: accession no. OMIX002324).
Acknowledgement
This research was funded by the Shandong Provincial Natural Science Foundation, China (ZR2020QC005), the National Natural Science Foundation of China (32272789), the National Natural Science Foundation of China (32000041), the Shandong Edible Fungus Agricultural Technology System (SDAIT-07-02),the Shandong Provincial Key Research and Development Plan(2021ZDSYS28), and the Qingdao Agricultural University Scientific Research Foundation (6631120076).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://doi.org/10.26599/FSHW.2022.9250144.

- 食品科学与人类健康(英文)的其它文章
- Betalains protect various body organs through antioxidant and anti-inf lammatory pathways
- Effects of Maillard reaction and its product AGEs on aging and age-related diseases
- Characterization of physicochemical and immunogenic properties of allergenic proteins altered by food processing: a review
- Polyphenol components in black chokeberry (Aronia melanocarpa)as clinically proven diseases control factors—an overview
- Food-derived protein hydrolysates and peptides: anxiolytic and antidepressant activities, characteristics, and mechanisms
- Recent advances in the study of epitopes, allergens and immunologic cross-reactivity of edible mango