
3-Epi-betulinic acid 3-O-β-D-glucopyranoside (eBAG) induces autophagy by activation of AMP-activated protein kinase in hepatocellular carcinoma
2024-01-24 01:11MengjieLiuXuqingLiuKiyueHeYongpingJinYujiLiJinrongGuoJingyuYngZhixingXuWenyiKng
食品科学与人类健康(英文) 2024年3期
Mengjie Liu, Xuqing Liu, Kiyue He, Yongping Jin, Yuji Li,Jinrong Guo, Jingyu Yng, Zhixing Xu,, Wenyi Kng
a School of Life Sciences, Henan University, Kaifeng 475004, China
b National R & D Center for Edible Fungus Processing Technology, Henan University, Kaifeng 475004, China
c Joint International Research Laboratory of Food & Medicine Resource Function, Kaifeng 475004, China
d Shenzhen Research Institute of Henan University, Shenzhen 518000, China
Keywords: eBAG Autophagy ATP AMPK Hepatocellular carcinoma
ABSTRACT 3-Epi-betulinic acid 3-O-β-D-glucopyranoside (eBAG) is a pentacyclic triterpene mainly distributed in food and medicinal plants, which exhibits various pharmacological properties. However, whether these functions are attributed to eBAG or additional components in these plants remain unknown. Herein, we report that eBAG exerted an inhibitory activity against hepatocellular carcinoma and esophageal cancer cells. EBAG induced non-apoptotic cell death in hepatocellular carcinoma cells. The eBAG-induced cell death was inhibited by knock-down of autophagy related gene (ATG) 5 and ATG7, by administration of 3-methyladenine,a selective autophagy inhibitor that suppresses phosphoinositide 3-kinase (PI3K), and by chloroquine, a classic autophagy f lux inhibitor. We demonstrated that eBAG induced an autophagy-mediated cell death. Application of eBAG mimicked cellular bioenergetics depletion leading to the reduction of intracellular ATP, activation of AMP-activated protein kinase (AMPK), and inhibition of mTOR. Co-treatment with compound C, an AMPK inhibitor, abrogated cell death induced by eBAG. We further validated the anti-tumor effect of eBAG in the murine xenograft model of hepatocellular carcinoma and found that eBAG treatment promoted the induction of autophagy and reduction of tumor growth in mice. As a functional food ingredient, eBAG is a potential therapeutic agent for the treatment of hepatocellular carcinoma and esophageal cancer.
1. Introduction
Primary liver cancer is still a major problem in all healthcare systems worldwide. Hepatocellular carcinoma (HCC) accounts for about 90% of non-metastatic liver tumors[1]. To improve the prognosis of patients with HCC, a variety of new drugs have been developed.Sorafenib, a first-line targeted treatment for advanced liver cancer,has been validated and compared with multiple drugs administrated in the later stage of HCC. Sunitinib[2], brivanib[3], cediranib[4], linifanib[5],and dovotinib[6], are also evaluated in HCC. Sunitinib, a tyrosine kinase inhibitor, was originally approved for the treatment of primary kidney cancer (advanced renal cell carcinoma) and advanced primary liver cancer[7], but did not achieve the expected goal in any of them.Therefore, f inding new targeted drugs is of great signif icance for the treatment of patients with HCC.
Triterpenoid saponins, an important class of bioactive products,are primarily divided into tetracyclic and pentacyclic triterpenoid saponins, among which pentacyclic triterpenes are common[8]. It has been reported that pentacyclic triterpene saponins, such as araloside A,oleanolic acid, andL-ascorbic acid are widespread in common foods,including fruits, vegetables, herbs, and other plant-based foods[8-9].Triterpene saponins have a wide range of clinical applications,including anti-cancer effects, and their anti-cancer effect on cell cycle arrest and apoptosis have been widely studied[10-11]. Triterpenoids are a kind of important secondary metabolites of plants, showing more and more important pharmacological effects and they are often considered as food and nutritional plants[12]. In addition, triterpenoids also have strong pharmacological activities for the treatment of poisoning,liver injury, cancer, diabetes and inflammation[13]. 3-Epi-betulinic acid 3-O-β-D-glucopyranoside (eBAG) is a pentacyclic triterpene mainly distributed in food and medicinal plants, which exhibit various pharmacological properties, such as anti-tumor and anti-cancer effects and used in traditional Chinese medicine for the treatment of cold fever, sore throat, and rheumatoid arthritis[14]. Ginseng,Ganoderma lucidum, Semen ziziphi spinosae and other dual-use medicinal products are mainly composed of triterpenoid compounds, which share a similar structure as eBAG. Thus, the triterpenoid compound eBAG bears the potential to be developed as a health food and special functional food[15-18]. However, whether these functions are attributed to eBAG or additional components in these plants remain unknown.Terpenoids were reported to induce apoptotic programmed cell death[19]. Recent studies have shown that terpenoids are able to trigger additional forms of cell death, such as autophagy[20]. In the current study, we identified that eBAG could induce autophagic cell death in HCC cells.
Autophagy is a self-feeding catabolic pathway, which regulates the dynamics of cell energy balance by degrading intracellular misfolded proteins, damaged organelles and lipid droplets[21-22]. The autophagy pathway is driven by a group of autophagy related genes(ATGs) that contribute to the formation of a double membrane capsule called autophagosome and to the process of autophagymediated degradation[23-24]. Autophagy degrades monosaccharides,fatty acids and amino acids through catabolic reactions for the synthesis of essential proteins, and produces adenosine triphosphate(ATP) for the maintenance of cellular bioenergenetics. Autophagy is a main degradation program of cells, which is up-regulated in response to various stresses, including nutritional starvation. Autophagy is also involved in the removal of harmful cytoplasmic components(proteotoxic aggregates, damaged organelles and pathogens) to promote cell survival[25-26].
Autophagy is associated with the pathogenesis of a variety of diseases involving energy metabolism. The role of autophagy in tumorigenesis and tumor inhibition is not fully understood. Autophagy is thought to play a dual role in cancer. Enhanced autophagy may prevent tumorigenesis by inhibiting chronic tissue damage,inflammation, and genomic instability, whereas autophagy may play a positive role in maintaining tumor metabolism, growth and survival through nutrient cycling[27-28]. When the energy in the form of ATP is limited, AMP-activated protein kinase (AMPK) is activated, which drives autophagy. Similarly, the lack of growth factors and/or amino acids leads to the inhibition of the mammalian target of rapamycin complex 1 (mTORC1), which regulates cell growth and metabolism via modulating protein and lipid synthesis, lysosome biogenesis, and autophagy. p62 is a mitochondrial protein degraded by autophagy through its LIR domain that binds to LC3 on the membranes of autophagosomes[29]. Depletion of p62 reduces liver injury and hepatocarcinogenesis caused by autophagy deficiency, indicating that the suppression of autophagy and/or aberrant accumulation of p62 could be the mechanism for liver cell transformation[30]. In this case,autophagy plays a suppressive role in tumorigenesis.
AMPK is a canonical regulator of energy balance and metabolism at the whole organism level[31]. AMPK is now considered to be a key cellular energy sensor, which regulates several processes including glycolysis, protein synthesis, lipid biosynthesis, and autophagy[32].After glucose withdrawal, AMPK is activated by the increase of intracellular AMP/ADP: ATP ratio. Activated AMPK phosphorylates downstream proteins to inhibit energy consuming anabolic pathways and initiate catabolic processes, such as autophagy[23]. AMPK inhibits mTORC1 by phosphorylating its raptor subunit (on ser722 and ser792) or activating the TSC complex[33-34]. In addition to inhibiting mTORC1, AMPK also promotes autophagy by directly phosphorylating and activating the ULK1 complex[35].
In this study, we compared the inhibitory effects of eBAG on a variety of tumor cell lines and found that eBAG could selectively inhibit the growth of tumor cells, especially HCC cells. We demonstrated that eBAG destroys cell energy balance, activates AMPK, and hence suppresses mTOR, which leads to the induction of autophagy. In addition, its application in murine xenograft model of HCC was also tested. We confirmed the induction of autophagy and suppression of tumorigenesis in eBAG-treated mice. Thus, eBAG may have the prospect of being developed as a health and functional food, and thus provide a new way to treat and/or prevent cancer.
2. Material and methods
2.1 General and isolation of eBAG
D-NMR spectra were recorded on a Bruker-AM-400 spectrometer(Billerica, MA, USA) tetramethylsilane (TMS) used as an internal standard. Electron ionization-mass spectrometry (EI-MS) spectra were obtained from a Thermo Trace DSQ II-mass spectrometer (Thermo Fisher Scientific Inc., MA, USA). Sephadex LH-20 (Pharmacia,Sweden) and silica gel (100–200, 200–300 mesh: Qingdao Marin Chemical Co., Ltd., Qingdao, China) were also used for the identification of eBAG. Isolation of eBAG was previously reported by our group[14].
2.2 Extraction and isolation
TheN-butanol extract (0.5 kg) was subjected to silica gel(200–300 mesh) column chromatography and eluted with CH2Cl2-MeOH gradient (50:1 to 1:1,V/V) to give 4 fractions (BuFr.1–BuFr.4). BuFr.4 was further subjected to fractionated on Sephadex LH-20 (100% MeOH), and then subjected to silica gel (H) column chromatography, eluted with CH2Cl2-MeOH gradient (8:1,V/V) to yield eBAG (20.0 mg)[14]. eBAG (molecular weight: 641 g/mol)was dissolved in DMSO. The purity of eBAG is more than 95%(Supplementary Figure).
2.3 Antibodies and chemicals
Antibody against p62 and Ki67 were purchased from Abcam(Boston, MA, USA). Antibodies against P-S6-S235/236, S6, mTOR,P-mTOR-S2448, P-AMPKα-T172, AMPKα1, cleaved PARP(Asp214), and P-ACC (ser79) were purchased from Cell Signaling Technology (Danvers, MA, USA). Beclin 1 antibody was purchased from Novus Biologicals (Littleton, CO, USA). Antibodies against LC3, Atg5, and Bax were purchased from Protein Tech (Wuhan,China). Atg7 antibody was purchased from Santa Cruz Biotechnology(CA, USA). zVAD-fmk and 3-methyladenine (3-MA) were purchased from AbMole (Shanghai, China). Ferrostatin-1, necrostatin-1,chloroquine, compound C, and rapamycin were purchased from MCE (Shanghai, China). Cell cycle and apoptosis detection kit was purchased from US EVERBRIGHT (Suzhou, China). Methylpyruvate(MP) was purchased from Sigma-Aldrich (Milwaukee, Germany).
2.4 Cell lines and DNA transfection
SNU739, KYSE30, and HCT116 were grown in RPM1640 medium containing 10% fetal bovine serum (FBS) in a humidified incubator containing 5% CO2at 37 °C. To establish SNU739/GFPLC3 stable cell line, proliferating SNU739 cells were infected with lentiviruses expressing Lv-GFP-LC3. After the transduction for 48 h,positive stable clones were selected with puromycin (2.5 μg/mL) for 2 weeks and the survived clones were combined and passaged. The RFP-GFP-LC3 plasmids were transiently transfected into SNU739 for 48 h, and fluorescence images were obtained using inverted fluorescence microscope. Lipofectamine 2000 (Invitrogen) was used for the transfection.
2.5 siRNA
A pool of two siRNA duplexes targeting human Atg5(forward 5’-CCAUCAAUCGGAAACUCAUTT-3’, reverse 5’-AUGAGUUUCCGAUUGAUGGTT-3’ and Atg7(forward 5’-GCCUCUCUAUGAGUUUGAATT-3’, reverse 5’-UUCAAACUCAUAGAGAGGCTT-3’) were purchased from Gene Pharma (Shanghai, China) and transfection was performed according to the manufacturer’s protocol.
2.6 Measurement of ATP
ATP levels were measured by Shanghai Enzyme-linked Biotechnology Kit (Shanghai, China). Linear regression curve was drawn based on the concentration of the standard and was used for calculating the concentration of each sample. The measurement was performed following the manufacturer’smanual.
2.7 Cell viability and cell death assay
Cell viability was measured by using Cell Counting Kit-8 (CCK-8)from Biosharp (Guangzhou, China). 10 uL of CCK8 solution was added to each well in a 96-well plate. Cells were incubated at the CCK8 solution at 37 °C for 2 h before the end of the experiment.The absorbance value at 450 nm was determined with a microplate reader. The cytotoxic activity of eBAG against SNU739 HCC cells,KYSE30 human esophageal squamous cell carcinoma cells and HCT116 human colorectal cancer cells were evaluated using CCK8.Cell death was measured by trypan blue (Solarbio) assay. The trypan blue staining solution was diluted with PBS to a final concentration of 0.4%. The living and dead cells were counted after cell suspension and 0.4% trypan blue staining solution were evenly mixed at 9:1.
2.8 Flow cytometric analysis of cell cycle
Cell-cycle distribution was analyzed by PI (Calbiochem) staining.Cells were fixed with 70% (V/V) ethanol for at least 2 h and incubated with 50 μg/mL PI for 30 min. The PI signal was measured by flow cytometry.
2.9 Immunofluorescence and fluorescence microscope
P62 fluorescent images were acquired with a laser scanning confocal microscope (ZEISS, LSM980, Germany) and analyzed with ZEN ImageJ software. GFP-LC3 cell images were acquired with a inverted fluorescence microscope (ZEISS Axio Observer7, Germany).
2.10 Immunoblotting
Cells were harvested, lysed in RIPA buffer (Solarbio) containing protease inhibitors and phosphatase inhibitors (Roche Applied Science), and proteins were quantified using the BCA Protein Assay Kit (Solarbio). Protein samples (15 μg each) were loaded to 10%–12%SDS-PAGE and were transferred to PVDF membrane (Millipore).The membranes were blocked with 5% bovine serum albumin (BSA)and probed with primary and second antibodies.
2.11 Electron microscopy
Cells were fixed with 2% glutaraldehyde in 0.1 mol/L phosphate buffer (pH 7.4), followed by 1% OsO4for 2 h in 4 °C. After dehydration,thin sections were stained with 1% uranyl acetate and lead citrate for observation with an electron microscope (Tecnai G2, FEI).
2.12 Animal experiments
Five-week-old BALB/c-nu/nu female mice were purchased from SPF Biotechnology Co., Ltd. (Beijing). All animal procedures were approved by the institutional animal care and use committee (IACUC)at Henan University, China and complied with its guidelines. Mice were randomly assigned to subcutaneous injection with SNU739 cells(5 × 106in 0.1 mL RPMI 1640) (n= 14). Tumor size was measured every five days. Tumor volume was calculated using the formula:Volume = (Width2× Length)/2. EBAG solution (20 mg/kg in PBS) or PBS was injected into the mice by i.p. every three days and mice were euthanized three weeks later. Tumor tissue samples were detected by immunohistochemistry.
2.13 Immunohistochemistry
After dewaxing, 5-μm tumor tissue sections were hydrated in gradient ethanol. For antigen retrieval, the slides were immersed in 0.01 mol/L sodium citrate buffer, heated for 7.5 min in a microwave oven at high heat and 7.5 min in a medium high heat. After blocked with goat serum, the slices wereincubated with antibodies against AMPKα-T172 (1:100), P-S6-S235/236 (1:500), P-ACC (ser79)(1:500), or Ki67 (1:200) for overnight at 4 °C. The slides were then incubated with biotinylated secondary antibodies and developed using peroxidase streptavidin and 3,3’-diaminobiphenylamine tetrahydrochloride (DAB) for 1 min. The nuclei were finally stained with hematoxylin[36].
2.14 Statistical analysis
All statistic analyses were performed using GraphPad Prism software (version8). The quantitative data obtained from experiments with biological replicates are presented as mean ± SD. Twotailed Student’st-test was used to analyze the quantitative data andPvalues < 0.05 were considered statistically significant.
3. Results
3.1 EBAG inhibits proliferation and promotes cell death in multiple cancer cell lines
Triterpenoids were previously reported to possess an antihepatocellular carcinoma effect[37]. EBAG is a triterpenoid we isolated previously. The structure of eBAG was determined based on the spectroscopic analysis (Fig. 1a). MS results of eBAG purity are shown in Fig. S1. Compounds with a similar structure of eBAG were found in multiple foods[38-39]. To determine the role of eBAG in the growth of HCC, we detected the proliferation and death of HCC SNU739 cells under the treatment of eBAG. SNU739 cells were exposed to various concentrations of eBAG, and cell viability was determined using a CCK8 kit. The results showed that SNU739 cells treated with eBAG for 48 h bore a markedly low viability with an IC50of 25 μmol/L for the compound. eBAG inhibited SNU739 cells in a dose-dependent manner (Fig. 1b). Triterpenoids reduce proliferation and promote death in multiple types of cancer cells[40]. To determine whether eBAG-regulated cell proliferation and death are generalized,we detected cell viability and cell death in human colon cancer HCT116 and esophageal cancer KYSE30 cells under the treatment of eBAG for 48 h. Similarly, exposure to eBAG markedly reduced the survival and enhanced the death of HCT116 and KYSE30 cells(Figs. 1c, d). Taken together, our data suggest that treatment of eBAG promotes cell death and decreases cell survival in multiple cancer cells.

Fig. 1 EBAG inhibits proliferation and promotes cell death in multiple cancer cell lines. (a) The chemical structure of eBAG. (b) Cell viability was detected by CCK8 and cell death was detected by trypan blue exclusive assay in SNU739 cells exposed to different concentrations of eBAG for 48 h. (c-d) Cell viability and cell death in (c) HCT116 and (d) KYSE30 cells treated with eBAG for 48 h. Cell viability and cell death were detected as described in (b). P values were determined by unpaired t-test. *P < 0.05; **P < 0.01; ***P < 0.001 (n = 3).
3.2 EBAG induces non-apoptotic, non-necrotic, and nonferroptic cell death
To define the cell death induced by eBAG, cell apoptosis,necroptosis, and ferroptosis were tested in SNU739 and/or KYSE30 cells. We pretreated the SNU739 and KYSE30 cells with benzyloxycarbonylvalyl-alanyl–aspartic acid (O-methyl)–fluoromethylketone (zVAD-fmk), a pan-apoptotic inhibitor, and determined cell viability and cell death under the treatment of eBAG. Cells treated with eBAG in the presence of zVAD-fmk for up to 48 h still underwent cell death (Figs. 2a-c, S1a), although the extent of cell death in zVAD-fmk-treated cells was less than that observed in cells treated with eBAG alone. After the treatment of eBAG, cells became rounded, irregular and then ballooned (data not shown). To determine whether cell death induced by eBAG is mediated by necroptosis or ferroptosis, we treated SNU739 cells with eBAG in the presence or absence of necroptosis inhibitor necrostatin-1 or ferroptosis inhibitor ferrostatin-1. We found that treatment of eBAG induced cell death regardless the presence or absence of necrostatin-1 or ferrostatin-1(Figs. 2d-f, S1b-c). In addition, expression of Bax, an apoptosis promoting protein, was no changed significantly under treatment of eBAG, supporting our observation that apoptosis is not the main causitive reason for eBAG-induced cell death (Figs. 2g, S1d).The proportion of cells in G0/G1 phase was increased due to the exposure to eBAG. In contrast, the proportion of S-phase cells in eBAG treatment group was lower than that in the control group (Fig. 2h).Collectively, our data indicate that treatment of eBAG leads to nonapoptotic, non-necrotic, or non-ferroptic cell death.
3.3 EBAG induces autophagy
Autophagic cell death is another type of programmed cell death,which plays an important role in cellular sensitivity to radiochemical therapy and other stresses[41]. We determined whether eBAG-induced non-apoptotic cell death could result from autophagy. To visualize autophagy, SNU739/GFP-LC3, a stable cell line expressing green fluorescent protein (GFP)-tagged LC3 was established. During the autophagic process, LC3 is concentrated in autophagosomes forming cytosolic punctate fluorescence, which serves as an indicator of autophagy. As shown in Fig. 3a, after being treated with eBAG for 48 h, SNU739 cells displayed a redistribution of GFP-LC3 from diffuse cytoplasmic location to discrete vesicular structures (punctate fluorescence). With the prolongation of exposure to eBAG, cells with punctate GFP-LC3 fluorescence increased in a time-dependent manner(Fig. S2a). Redistribution of LC3 was confirmed biochemically by western blot. Intracellular LC3 of eBAG-treated cells underwent a conversion from the LC3-I isoform to the LC3-II isoform, which is specific for autophagosomes/autolysosomes (Figs. 3b, S2b). p62 is a mitochondrial protein, which is degraded by autophagy. The expression of p62 decreased strikingly following the treatment of eBAG in cells (Figs. 3c, S2c-d). Immunofluorescent staining also showed that mitochondrial localization of p62 was strikingly reduced after eBAG treatment (Fig. S2e), supporting the notion that enhanced autophagy in eBAG-treated cells promoted the degradation of p62.Electron microscopy of eBAG-treated SNU739 cells showed the formation of double- or multi-membrane-bound structures containing recognizable cellular organelles and high electron-density substances,characteristics of autophagosomes and autolysosomes (Fig. 3d).
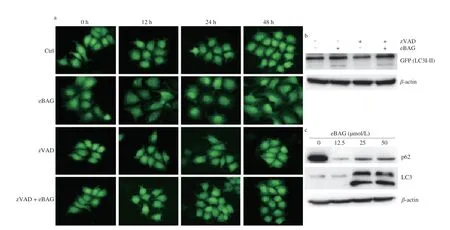
Fig. 3 EBAG induces autophagy in HCC cells. SNU739/GFP-LC3 cells were pretreated with zVAD-fmk (20 μmol/L) for 2 h, followed by eBAG (25 μmol/L)treatment for 0 to 48 h. (a) Punctate GFP-LC3 fluorescence in SNU739/GFP-LC3 stable cells. (b) Expression of GFP-LC3-I and -II in SNU739/GFP-LC3 stable cells treated with/without eBAG and/or zVAD-fmk. (c) Expression of p62 and LC3 in different concentrations of eBAG in SNU739 cells. (d) Electron micrograph of SNU739/GFP-LC3 cells treated with eBAG for 48 h. White arrows indicated the autophagosomes/autolysosomes. (e) SNU739 cells were transiently transfected with RFP-GFP-LC3 plasmid and treated with/without eBAG. After 48 h, red fluorescence (RFP), green fluorescence (GFP) and yellow fluorescence (merge) were detected.
In the acidic environment where autophagosome and lysosome fuse, GFP is easily degraded, and the fluorescence will be quenched[42].In order to observe the alteration of autophagy flux induced by eBAG,we transfected RFP-GFP-LC3 plasmid into SNU739 cells, and the live cells were photographed with confocal fluorescence microscope.Co-expression of RFP- and GFP-LC3 in autophagy precursors was yellow. After the appearance of autophagosome, GFP signal was weakened, but RFP signal still existed. No over accumulation of RFP-GFP-LC3 (yellow) or RFP-LC3 was observed, indicating that eBAG possibly upregulates the autophagy formation, but not alters autophagy flux (Fig. 3e). Together, our data suggest that eBAG treatment induces autophagy in cancer cells.
3.4 Suppression of autophagy abrogates eBAG-induced nonapoptotic cell death
To detect the role of autophagy in eBAG-induced non-apoptotic cell death, we pretreated the cells with chloroquine (CQ) for 6 h and then treated them with eBAG. CQ is an autophagy inhibitor,which weakens the acid environment of lysosome, eliminates the enzyme activity of lysosome, and hence inhibits autophagy-mediated degradation. It was found that eBAG-induced morphological alterations of cells, such as rounding and detachment, were completely suppressed by CQ. EBAG-treated cells showed increased viability and decreased death due to the presence of CQ (Fig. 4a). 3-Methyladenine(3-MA), an inhibitor of class III PI3-Kinase, is a cell-permeable autophagic sequestration blocker. Pretreatment of 3-MA led to a downregulation of autophagosome/autolysomes in the eBAG-treated cells, supporting the notion that eBAG promotes the formation of autophagy, but not the fusion between autophagosome and lysosome.Moreover, treatment of 3-MA abrogated eBAG-induced cell death and promoted cellular survival (Fig. 4b).
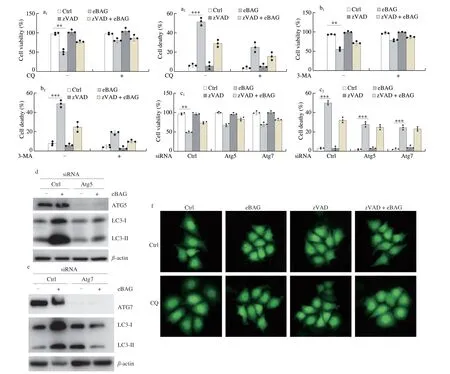
Fig. 4 Inhibition of autophagy alleviates eBAG-induced cell death. (a-b) SNU739/GFP-LC3 cells were pretreated with CQ (10 μmol/L) or 3-MA (25 μmol/L) for 6 h. zVAD-fmk and eBAG were added as described in Figs. 2 and 3. Cell viability and cell death assays followed the protocols described in materials and methods.Data are shown as mean ± S.D. (n = 3). (c) SNU739 cells were treated with siRNA of ATG5 or ATG7 for 24 h, and then eBAG (25 μmol/L) and/or zVAD-fmk (20 μmol/L)were added continuously for additional 48 h. Cell viability and cell death were detected. (d-e) Expression of ATG5/ATG7 and GFP-LC3 in ATG5/ATG7 siRNA SNU739 cells. (f) Punctate GFP-LC3 fluorescence in stable SNU739/GFP-LC3 was detected after treatment with eBAG for 48 h in the presence/absence of CQ.
To validate the role of autophagy in non-apoptotic cell death induced by eBAG, we explored the effects of autophagy-related gene-5(ATG5) and ATG7 depletion by gene silencing on eBAG-induced cell death. Small interfering RNA (RNAi) targeting ATG5 and ATG7 markedly reversed the effects of eBAG on cell viability and cell death(Fig. 4c)and reduced the endogenous expression of ATG5 and ATG7(Figs. 4d-e, S3a). During the eBAG treatment, a large fraction of CQ(Figs. 4f, S3b) and 3-MA (Figs. S3c-d) treated cells appeared healthy and the formation of autophagosomes was markedly suppressed.Together, we conclude that the non-apoptotic cell death induced by eBAG is related to autophagy.
3.5 EBAG-induced cellular energy deficiency contributes to autophagy
To elucidate the molecular mechanisms by which eBAG induces autophagy, we determine whether eBAG directly affects the expression of autophagy-associated genes. We detected the expression of beclin 1 (ATG6) firstly. Beclin-1 is a protein that interacts with either BCL-2 or PI3k class III, playing a critical role in the regulation of both autophagy and cell death. As shown in Figs. 5a and S4a, SNU739/GFP-LC3 cells treated with eBAG and/or zVAD-fmk showed no accumulation of beclin 1. The essential function of autophagy is to meet the needs of metabolism. Energy supply is important for the regulation of autophagy. Thus, we asked whether eBAG induces autophagy by affecting cellular energy. To test this possibility, we measured ATP levels in cells treated with eBAG. SNU739 cells treated with eBAG showed a time-dependent decrease in levels of intracellular ATP (Fig. 5b), suggesting that cellular energy depletion might be responsible for the eBAG-induced autophagy. Thus, we next determined whether the depletion of ATP production and autophagy induction in the eBAG-treated cells could be reversed by cotreatment with a cell-permeable form of pyruvate,methylpyruvate (MP), which can be oxidized in the tricarboxylic acid cycle to produce NADH that fuels the electron transport and ATP production. We found that MP treatment significantly increased eBAG-induced cell viability (Fig. 5c). MP restored ATP production in eBAG-treated cells to levels that were close to those observed in untreated cells (Fig. 5d). The development of autophagosomes/autolysosomes was inhibited after addition of MP (Figs. 5e, S4b). In conclusion, our data suggest that eBAG-induced cellular bioenergetics destruction contributes to the formation of autophagy and mediates non-apoptotic cell death.

Fig. 5 EBAG induces cellular energy deficiency leading to autophagy. (a) Expression of beclin 1 in SNU739/GFP-LC3 cells treated with eBAG (25 μmol/L) and/or zVAD-fmk (20 μmol/L) for 48 h. (b) ATP levels of cells grown in 25 μmol/L eBAG for 0–8 h. (c) Cell viability in cells treated with methylpyruvate (MP) and/or eBAG for 48 h. (d) ATP levels of cells grown in the presence or absence of MP and/or eBAG. (e) Autophagosomes/autolysosomes in MP- and/or eBAG-treated SNU739/GFP-LC3 cells. Data represent average of 3 independent experiments ± S.D. *P < 0.05; **P < 0.01; ***P < 0.001 (n = 3).
3.6 EBAG induces autophagy by activating AMPK
Previous studies have shown that reducing ATP levels induces autophagy in the eBAG-treated cells (Fig. 5b). AMPK is a cell energetic sensor. Thus, we asked whether eBAG treatment triggers the activation of AMPK. We measured the phosphorylation of AMPK. As intracellular AMP/ATP ratios rose, AMPK is activated.We found that eBAG treatment resulted in enhanced phosphorylation of AMPK at Thr-172, an active status of AMPK. Furthermore,the eBAG treatment also led to the inhibition of mTOR and S6 activation, downstream targets of AMPK. The phosphorylation of mTOR and S6 decreased substantially after eBAG treatment(Figs. 6a-b, S5a-b). In addition, we also found that the phosphorylation of AMPK increased in a time-dependent and concentration dependent manner (Figs. S5c-e). To determine whether AMPK activation plays a direct role in eBAG-induced autophagy, we detected the capability of eBAG to induce autophagy in cells treated with AMPK inhibitor(compound C, CC). AMPK inhibitor CC suppressed eBAG-induced AMPK phosphorylation (Fig. 6a), reduced eBAG-triggered cell death, increased cell viability (Fig. 6c) and decreased autophagy(Figs. 6d, S5f). In addition, we found that eBAG displayed a similar effect on the regulation of AMPK-mTOR-S6 signaling as rapamycin(mTOR inhibitor) did (Figs. 6e, S5g). These data suggest that AMPK and its downstream targets are important mediators of eBAG-induced autophagic cell death.

Fig. 6 EBAG induces autophagy by activating AMPK. (a) Expression of p-ACC (ser79), phospho-AMPK (T172), AMPK, phospho-mTOR (S2448), mTOR,phospho-S6 (S235/236), and S6 in SNU739/GFP-LC3 cells treated with 25 μmol/L eBAG and/or 10 μmol/L compound C for 48 h. (b) Concentration-dependent effects of eBAG on the activation of AMPK. SNU739 cells were treated with 30, 50, and 70 μmol/L eBAG for 48 h and then detected by Western blotting. (c) Cell death and cell viability in eBAG and/or compound C (CC)-treated SNU739 cells. Data are shown as mean ± S.D. (n = 3). (d) Punctate GFP-LC3 fluorescence in SNU739/GFP-LC3 cells treated with eBAG in the presence or absence of compound C. SNU739/GFP-LC3 cells were pretreated with 10 μmol/L compound C for 8 h followed by treatment with 25 μmol/L eBAG for 48 h. (e) SNU739 cells were exposed to eBAG (25 μmol/L) and/or rapamycin(20 nmol/L)for 48 h.Western blotting was used for the detection of AMPK-mTOR-S6 signaling molecules.
3.7 EBAG inhibits HCC tumor growth
To evaluate the effect of eBAGin vivo, we detected the impact of eBAG on tumor growth in xenograft murine tumor model. SNU739 cells were inoculated subcutaneously in nude mice. Five days after the inoculation, 20 mg/kg eBAG in PBS was injected into the mice by i.p. every 3 days, and at the end, tumors were harvested after the inoculation for 3 weeks (Figs. 7a, b). The volume and weight of tumors in the eBAG treated mice were much smaller (Fig. 7c)and lighter (Fig. 7d) as compared with those in PBS-treated mice. We performed an immunohistochemical staining on tumors collected and found that p-AMPK and p-ACC were markedly increased, whereas p-S6 and Ki67 were decreased in tumors from mice treated with eBAG(Fig. 7e). These data further support our notion that eBAG suppresses tumor growth through regulating AMPK-mTOR-S6 signaling.

Fig. 7 EBAG inhibits HCC tumor growth. Nude mice (n = 28) were inoculated subcutaneously with 5 × 106 SNU739 cells. Five days after the inoculation, the mice were randomly divided into 2 groups treated with 20 mg/Kg eBAG or PBS by i.p. every 3 days. Tumors were collected for assays 3 weeks later.(a) Schematic diagram of animal experiment. (b) Images of tumors from mice treated with or without eBAG. The tumors shown here are representatives of three repeated experiments. (c) Tumor weight in mice treated with or without eBAG (n = 14). (d) Tumor volumes in mice treated with or without eBAG (n = 14).(e) Immunohistochemical (IHC) staining of tumor tissue sections.
4. Discussion
Multiple triterpenoids, such as betulinic acid, have been characterized to bear antitumor activities[43-44]. Researches showed that pentacyclic triterpenes isolated from functional foods and additives display anti-tumor and anti-inflammation effects and used for the treatment of cold fever, sore throat, and rheumatoid arthritis, and cancers. In the current study, we characterized the tumor suppressive effect of eBAG, a pentacyclic triterpene we isolated recently, on the growth of a variety of tumor cells (Fig. 1). We validated the suppressive role of eBAG in HCC, colorectal cancer, and esophageal cancer and explored the mechanisms by which eBAG induced cell death of HCC. We showed that eBAG-induced cell death was not due to apoptosis, necroptosis, and ferroptosis. Instead, we demonstrated that eBAG treatment induced the formation of autophagosome. The cell death resulted from the eBAG treatment was blocked by the addition of CQ and 3-MA, two autophagy inhibitors. Silencing of Atg5 and Atg7 also inhibited autophagy formation and markedly reduced the eBAG-associated cell death. These data strongly support the conclusion that eBAG induces autophagic cell death.
Cellular bioenergetics deficiency is a critical trigger for autophagy induction[45-46]. The elevation of AMP/ATP ratio in cells activates cellular energetic sensor, AMPK, leading to the suppression of anabolism and promotion of catabolism[47-50]. We identified that eBAG treatment depleted cellular bioenergetics and resulted in AMPK activation. Inhibition of AMPK with compound C, an AMPK inhibitor, significantly reduced the autophagic cell death induced by eBAG. Furthermore, the administration of eBAG to nude mice substantially increased the level of AMPK phosphorylation, decreased S6 phosphorylation, and suppressed xenograft tumor growth. These phenotypes clearly indicate that eBAG induces autophagic cell death by activating AMPK, leading to the suppression of mTOR-S6 signaling, which contributes to antitumor activity of the agent (Fig. 8).
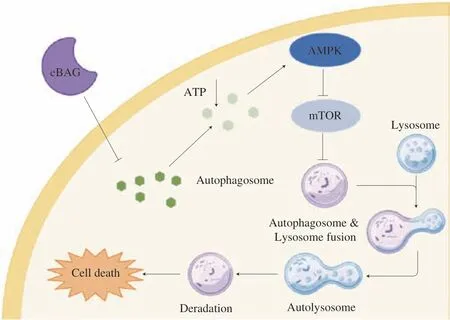
Fig. 8 Role of eBAG in autophagy. Schematic diagram depicting the action of eBAG in the induction of autophagy.
The most common forms of programmed cell death are apoptosis,autophagy, necroptosis, and ferroptosis[51-53]. To elucidate which programmed cell death is involved in eBAG-induced cell death, we treated SNU739 cells with different dosages of eBAG, and measured cellular apoptosis, necrosis, autophagy, and ferroptosis. Our data showed that eBAG induced autophagic cell death, but not apoptosis,necroptosis, or ferroptosis. A recent report showed that ferroptosis is an intracellular iron-dependent form of cell death that is distinct from apoptosis, necrosis, and autophagy[54]. Ferroptosis could trigger an autophagy dependent form of cell death[55]. Although we observed a well-defined autophagic cell death in the eBAG-exposed cells in the current study, we were unable to detect ferroptosis in the cells.Ferroptosis occurs normally with the accumulation of ROS and oxidation of lipid. The elevated autophagy aids in the removal of ROS and degrades lipid droplets in cells, which may partially explain why the eBAG-treated cells bear a high level of autophagy, but no ferroptosis.
In conclusion, in the current study, we characterized the functions of a lupine pentacyclic triterpenoid, eBAG,and identified that the compound exerts an anti-tumor activity by inducing autophagic cell death through depleting of cell energy supply. Most importantly,we found that eBAG could induce cell death even if apoptosis,necroptosis and ferroptosis were suppressed, which means that eBAG bears a therapeutic potential in apoptosis-resistant cancers, in particular those in HCC and colorectal cancer.
Ethics statement
Protocols for animal usage were approved by the institutional animal care and use committee (IACUC) at Henan University,China. All animal experiments were conducted on the basis of the institutional guidelines, and were approved by the Laboratory Animal Center of Henan University.
Conflict of interests
Wenyi Kang is an associate editor forFood Science and Human Wellnessand was not involved in the editorial review or the decision to publish this article. The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
This work was supported by Henan Provincial Science and Technology Research Project (212102310355) and the National Natural Science Foundation of China (82020108024 and 32161143021).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://doi.org/10.26599/FSHW.2022.9250122.

- 食品科学与人类健康(英文)的其它文章
- Betalains protect various body organs through antioxidant and anti-inf lammatory pathways
- Effects of Maillard reaction and its product AGEs on aging and age-related diseases
- Characterization of physicochemical and immunogenic properties of allergenic proteins altered by food processing: a review
- Polyphenol components in black chokeberry (Aronia melanocarpa)as clinically proven diseases control factors—an overview
- Food-derived protein hydrolysates and peptides: anxiolytic and antidepressant activities, characteristics, and mechanisms
- Recent advances in the study of epitopes, allergens and immunologic cross-reactivity of edible mango