
用于高灵敏快速核酸检测的荧光碳点
2024-01-22 12:11常建桥许慧敏谢文菁张洋祁玲范楼珍李勇
物理化学学报 2023年12期
常建桥,许慧敏,谢文菁,张洋,祁玲,范楼珍,*,李勇
1北京师范大学化学学院,理论计算光化学与放射性药物教育部重点实验室,北京 100875
2中国医学科学院北京协和医学院内科,国家癌症中心/国家癌症临床研究中心/癌症医院,北京 100021
3中国医学科学院北京协和医学院胸外科,国家癌症中心/国家癌症临床研究中心/癌症医院,北京 100021
1 引言
PCR技术是指通过多种酶的作用,双链DNA在高温下变性为单链DNA。根据互补碱基配对原则,利用DNA聚合酶从单链DNA复制出新的DNA,实现DNA体外扩增1,2。逆转录PCR (RTPCR)是指当体系内核酸为RNA时,通过逆转录酶将RNA逆转录成cDNA,再进行体外扩增的过程。实时定量PCR (qPCR)是在PCR的基础上,向体系中加入染料或荧光探针,使扩增后的DNA具有荧光性,结合荧光信号强度与扩增增量的关系,可以在扩增过程中实时监测目的DNA的拷贝数3–6。自从新冠病毒流行以来,所采用的标准核酸检测技术为RT-qPCR技术,其将RT-PCR技术与qPCR相结合,尽管具有强大的检测性能,但其作为新冠病毒的诊断技术仍然存在一些局限性,例如需要相对较长的检测过程(2–4 h),专业的设备(热循环仪、PCR仪等)、实验室以及专业的操作人员等7–9。因此,开发出新冠病毒快速、灵敏的即时检测(POCT)是非常必要的。
快速抗原检测是一种针对特定抗原的POCT,其主要是基于横向流动免疫分析(LFIA),当含有抗原的样品滴入到样品垫后,流到结合物释放垫与抗体1结合,抗体1上携带有色颗粒(例如胶体金),抗原-抗体1-金复合物流向试纸条的检测区。检测区是一种多孔膜,存在测试线和控制线。当复合物经过测试线时,抗原与测试线上的抗体2结合,复合物留在测试线,金颗粒发生响应,显示阳性。而控制线上的响应则表明正确的液体流过试纸条。抗原检测可提供早期病毒感染的证据10,11,具有成本低、操作方法简单、检测时间短(20 min或更短)等优势12。但是其检测结果可能会受到其他具有相似结构的化学物质的影响,导致其灵敏度为75%–98%,特异性为95%–99%,容易出现假阳性结果13。由于病毒载量的影响,LFIA的检测限为1.12 × 105–3.57 × 106copys∙mL-114,只能在出现症状的第一周有效,容易导致假阴性结果15。抗原检测这种相对较低的灵敏度给检测带来了多重障碍,需要进一步研究以提高其灵敏度和准确性16–18。
碳点(CDs)是一种零维碳纳米材料,具有优异的光学性质、良好的水溶性、较好的生物相容性以及广泛的原料来源等诸多优点19–23,已经应用于检测24–26、诊断27–29以及药物的递送等领域30–32。本研究使用邻苯二胺和精氨酸作为前驱体,合成了一种基于胍基修饰的荧光碳点(GCDs)(如图1a)。当GCDs与分子信标(Beacon)通过氢键结合后,GCDs的荧光被Beacon所修饰的荧光基团淬灭。遇到目标核酸(Target DNA)之后,Target DNA通过碱基互补配对,与GCDs竞争Beacon分子,GCDs与Beacon脱离,其自身荧光恢复,根据荧光恢复的强度来判断体系内Target DNA的存在(图1b),基于这种灵敏的荧光“off-on”,可实现在5 min内快速准确的核酸检测,并可检测到体系内0.005 fmol∙L-1(约300 copys∙mL-1)的核酸序列,我们预计,新建立的COVID-19低成本无扩增检测将有助于开发新的核酸检测平台,以实现COVID-19和其他病原体的高灵敏度和高特异性的快速即时检验。

图1 (a) GCDs合成过程,(b) GCDs核酸检测机理Fig.1 (a) GCDs synthesis process and (b) nucleic acid detection mechanism of GCDs.
2 实验部分
2.1 实验材料和实验仪器
邻苯二胺和精氨酸购自上海阿拉丁生化科技股份有限公司,纯度为99%。二氯甲烷和甲醇纯度为99%,柱层析硅胶和中性三氧化二铝购自山西诺泰生物科技有限公司。磷酸盐缓冲液(PBS)、0.22 μm滤膜和7000 kD透析袋购自北京科龙生物科技有限公司。实验所使用的核酸序列购自上海生工生物有限公司,主要核酸序列见表1。

表1 主要核酸序列Table 1 Major nucleic acid sequence.
荧光光谱仪(法国Horiba Jobin Yvon公司,Fluorolog-3)、透射电子显微镜(美国赛默飞世尔科技公司,Talos F200S)、傅里叶变换红外光谱仪(日本岛津公司,IRAffinity-1)、X射线光电子能谱(美国Thermo Fisher公司,ESCALAB 250Xi)、拉曼光谱仪(法国Horiba Jobin Yvon,LabRAM)。
2.2 GCDs的合成
向60 mL去离子水中加入0.15 g邻苯二胺和0.3 g精氨酸,超声15 min使其完全溶解。将内胆置于高压反应釜中,180 °C反应5 h,反应结束后自然冷却。使用孔径为0.22 μm的水系滤膜对产物进行过滤。将过滤后的样品烘干,以甲醇:二氯甲烷(9 : 1)混合溶剂作为洗脱剂,使用200–300目的硅胶和中性氧化铝进行两次提纯,收集黄色荧光碳点。将得到的产物使用旋转蒸发仪去除有机溶剂后溶解在水中,得到GCDs溶液。
2.3 GCDs胍基数目的测定
应用坂口反应来对GCDs表面的胍基数目进行测定。取50 μL不同浓度的精氨酸溶液,向其中加入10 μL NaOH溶液,再加入10 μL α-萘酚溶液,混合均匀后加入10 μL 10%的次氯酸钠溶液,静置5 min后测量体系内500 nm处的吸收值,绘制精氨酸标准曲线,得到吸收光强度与精氨酸胍基数目的对应关系。按照相同的方法对GCDs中的坂口反应吸收值进行测量,对照标准曲线得到GCDs的胍基数目。
2.4 GCDs-Bea的制备
将GCDs与Beacon在PBS中以不同比例混匀后,涡旋震动10 s,室温避光静置15 min,然后使用7000 kD的透析袋进行透析2–3天,除去未与Beacon结合的游离GCDs,取出透析袋内液得到GCDs-Beacon (GCDs-Bea)。
使用JY-SPBT电泳设备对GCDs-Bea进行琼脂糖凝胶电泳分析。凝胶浓度2%,电压85 V,电泳时间40 min,Running Buffer为TAE缓冲液。电泳结束后,使用凝胶成像仪在254 nm激发波长下对琼脂糖凝胶进行成像,使用Image J软件对凝胶成像结果进行灰度值分析。
2.5 核酸检测
向2 mL GCDs-Bea中分别加入100 μL的PBS,Target DNA,Mismatch DNA等核酸序列,在室温混匀后使用荧光光谱仪测量体系内荧光光谱,并对570 nm处的荧光变化率(F-F0)/F0×100%进行分析。
3 结果与讨论
3.1 GCDs结构表征
GCDs的结构表征如图2所示,图2a为GCDs的TEM图像,GCDs具有均匀的分散状态,其平均粒径约为4 nm,大小均一,高分辨TEM图显示GCDs的晶格间距为0.21 nm,对应于石墨的(100)晶面的衍射。图2b为傅里叶变换红外光谱(FT-IR)结果表征,在3433 cm-1处有对应N―H的振动峰,表明GCDs表面存在―NH2基团。在3172 cm-1处有对应羧酸―OH的振动峰,表明GCDs表面存在―COOH基团,1506和1637 cm-1处有两个强峰,这是C=C/C=N的伸缩振动特征峰。通过坂口反应测定后,对应精氨酸浓度与吸收值的标准曲线,如图2c所示,计算表明一个GCDs上约带有3–4个胍基。为了进一步探究GCDs的结构,分别进行了X射线光电子能谱(XPS)和拉曼光谱的表征。如图2d所示,用XPS确定了GCDs的元素组成。将XPS数据拟合后得到高分辨谱图,如图2e的高分辨C 1s光谱显示,GCDs中碳元素主要由C=C/C―C (284.6 eV)、C―N/C―O (285.1 eV)和C=O (288.3 eV)形式构成。图2f拉曼光谱进一步表征了GCDs的结构,在1385 cm-1(D峰,碳材料中的无序态)和1597 cm-1(G峰,碳材料中的结晶态)附近有两个宽峰,分别对应碳原子的sp3和sp2杂化振动。GCDs的ID/IG约0.6,这表明所制备的GCDs具有高度的晶体结构。这一结果也与HRTEM结果一致。综上所述,GCDs是中心为较好共轭性的刚性碳核结构,边缘具有―NH2、―COOH以及3–4个―C3H4基团的荧光碳点(图1a)。

图2 GCDs的(a) TEM图,高分辨TEM图以及尺寸统计分布图,(b) FT-IR图,(c)坂口反应标准曲线,(d) XPS全谱,(e) C 1s高分辨谱图,(f)拉曼图Fig.2 (a) TEM,high resolution TEM and size statistical distribution diagram,(b) FT-IR,(c) standard Sakakuchi reaction curve,(d) XPS full spectrum,(e) C 1s high resolution spectrum,(f) Raman diagram of GCDs.
3.2 GCDs的光学性质
图3a是GCDs的紫外可见吸收光谱图,GCDs在波长为210 nm处存在明显的吸收峰,对应于sp2共轭结构的π–π*的跃迁吸收。在波长为258 nm附近存在几个较弱吸收峰,对应于C=O等结构的n–π*跃迁吸收。GCDs在415 nm处有特征吸收峰,对应的荧光发射峰位置为570 nm。图3a插图为GCDs在日光下的照片和紫外光照射下的荧光图像。使用积分球测得GCDs在410 nm 激发下的荧光量子产率为9.8%。图3b是GCDs的荧光激发光谱和发射光谱,GCDs的发射光谱从激发光波长400 nm增加到520 nm时,最大发射峰均在570 nm,不存在激发依赖现象,与图3c的二维荧光光谱图对应。并且GCDs室温存放17个月后,荧光发射位置不发生移动,荧光强度下降约4.5%,如图3d所示,GCDs的发光稳定性较好。
3.3 GCDs-Bea的制备
图4a为琼脂糖凝胶电泳阻滞实验结果,Beacon与凝胶中的GelRed结合后,在254 nm激发下产生荧光条带,经过凝胶成像系统拍照后,使用Image J软件测量条带灰度值,结果表明,向Beacon中加入GCDs后,Beacon亮度减弱,GCDs与Beacon发生相互作用,阻碍了Beacon条带的移动,当向Beacon中加入2倍量GCDs时,Beacon条带几乎消失(图4a左一),过量的GCDs与Beacon发生相互作用后完全阻滞了Beacon的移动。
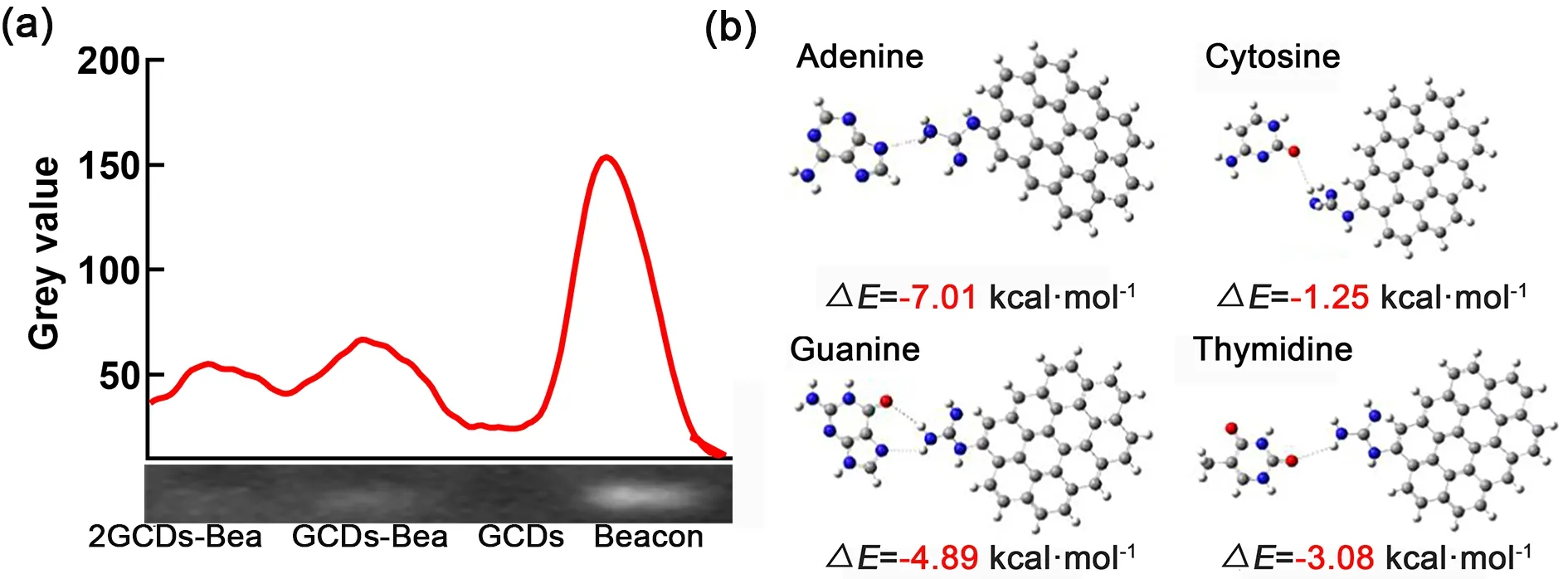
图4 GCDs与Beacon连接结果。(a)琼脂糖凝胶电泳条带的灰度值,(b) GCDs上胍基基团与Beacon四种碱基相互作用能量Fig.4 GCDs-Bea connection result.(a) Gray value of bands in agarose gel electrophoresis,(b) calculation results of guanidine groups and beacon bases.
对GCDs的结构进行简化后计算GCDs与Beacon的结合能。使用几何建模方法确定了GCDs中的氨基(―NH2)、羧基(―COOH)和胍基(―CN3H4)与腺嘌呤(A)、鸟嘌呤(G)、胞嘧啶(C)和胸腺嘧啶(T)四种碱基之间的相互作用,并在b3lyp/6-31g(d)水平上优化了几何结构,通过从头算量子化学方法计算了这些模型的相互作用能量33。如图4b的计算结果表明,胍基与Beacon的相互作用能量为-1.25 – -7.01 kcal∙mol-1(1 kcal∙mol-1=4.1868 kJ∙mol-1),而氨基和羧基与Beacon的相互作用能量相似(分别为0.58 – -2.21 kcal∙mol-1和2.55 – -3.07 kcal∙mol-1),表明胍基在与Beacon相互作用时具有明显的能量优势。
3.4 核酸检测结果的特异性与灵敏度
向GCDs-Bea (Control)中分别加入Target DNA以及错配DNA (Mismatch DNA,序列与Target DNA不同),结果如图5a所示。GCDs-Bea与Target DNA混匀后测量荧光光谱,荧光强度增强6倍,但Mismatch DNA组的荧光没有发生明显变化,显示GCDs-Bea核酸检测具有较强的特异性。为了明确检测方法的特异性,设置了55个样本,其中34个为含有不同浓度的Target DNA的阳性样本,21个含有不同序列的Mismatch DNA的阴性样本,使用GCDs-Bea对55个样本进行荧光测定并分析,检测阈值设置为荧光强度变化率为20%。结果如图5b,阳性样本平均荧光增长率为124.75%,阴性样本平均荧光增长率为0.04%。其中,阴性组存在一样本荧光增长率为20.35%,为假阳性结果。所以GCDs-Bea的核酸检测特异性=真阴性样本/(真阴性样本+假阳性样本) × 100% = 95.45%。
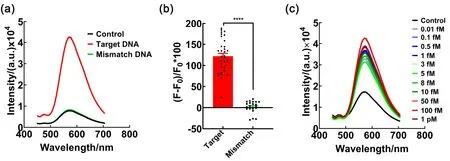
图5 GCDs-Bea核酸检测的(a)特异性,(b)多样本特异性,(c)灵敏度。图c中的浓度为检测时的起始浓度,终浓度为该浓度的5%Fig.5 (a) Specificity,(b) multiple sample,(c) sensitivity of GCDs-Bea nucleic acid detection. The concentration in the figure c is the initial concentration at the time of detection,and the final concentration is 5% of that concentration.
为了确定检测的灵敏度,将GCDs-Bea与不同浓度的Target DNA进行孵育,测量分析GCDs的荧光变化,图5c显示出GCDs-Bea对不同浓度的Target DNA产生不同的荧光现象,DNA浓度即使低至0.01 fmol∙L-1(终浓度约为300 copys∙mL-1)时,仍然有荧光增加现象,表明了GCDs-Bea具有较强的灵敏度。对比RT-qPCR检测方法的检测限2.59 × 102–1.04 × 103copys∙mL-1,快速抗原检测试剂盒的1.12 × 105–3.57 × 106copys∙mL-114,GCDs-Bea的检测方法可以灵敏地检测出目标核酸序列,而不需要进行核酸扩增步骤。此外,GCDs-Bea可以在两周内保持稳定,不影响检测效果,将DNA加入到GCDs-Bea后,可立即进行荧光测定,获得检测结果,该过程预计在5 min内。
但是在检测过程中发现,荧光恢复的强度与Target DNA的浓度不呈线性关系,推测原因与GCDs-Bea的连接机理有关。GCDs与Beacon连接的数量比并不一致,所以Target DNA与Beacon特异性结合后,脱离下来的GCDs数量不一致,恢复的GCDs的荧光有所不同,导致DNA浓度增大后,荧光值可能减小。但是可以确定的是,荧光值虽然减小,但是依然比Control组高20%以上。
3.5 其他核酸序列的检测
为了明确GCDs-Bea体系能否检测其他病毒或者疾病,将GCDs与不同序列的Beacon分子连接(Beacon2和Beacon3),然后将其与对应序列Target DNA2和Target DNA3进行测试,如图6a–c所示,更换Beacon核酸序列后,GCDs-Bea在检测到Target DNA后,GCDs的荧光强度变化率均在50%以上。将Target DNA的核酸序列改变一个碱基(Mis1)或4个碱基(Mis4),再分别与GCDs-Bea2反应,如图6d所示,当GCDs-Bea体系内存在Target DNA,荧光强度变化率为24%,改变Target DNA中的一个碱基,荧光强度变化率降为13%,改变4个碱基后,变化率仅为3%,与Mismatch DNA相同。检测体系具有与Beacon1相似的特异性和灵敏度。实验表明GCDs-Bea检测体系可以通过改变Beacon的核酸序列,进行不同病毒或疾病的核酸检测。

图6 GCDs和不同核酸序列Beacon2 (a)和Beacon3 (b)的灵敏度结果,(c) Beacon、Beacon2和Beacon3的荧光检测结果,(d) GCDs-Bea2的核酸检测的特异性,(e)混合样品的核酸检测。图中的浓度为检测时的起始浓度,终浓度为该浓度的5%Fig.6 The sensitivity results of GCDs and different nucleic acid sequences of Beacon2 (a) and Beacon3 (b),(c) fluorescence results of Beacon,Beacon2 and Beacon3,(d) specificity of nucleic acid detection of GCDs-Bea2,(e) the nucleic acid detection of mixed samples.The concentration in the figure is the initial concentration at the time of detection,and the final concentration is 5% of that concentration.
保持核酸终浓度一致,将不同序列的核酸进行混合后,GCDs-Bea依然能检测到其中含有的微量Target DNA,如图6e所示,MisDNA2、3、4分别为改变Target DNA中的2、3、4个碱基,T +Mis2 + 3 + 4为Target DNA、MisDNA2、MisDNA3和MisDNA4混合物。0.25T为0.25倍的0.01 fmol∙L-1Target DNA,即向体系内加入0.0025 fmol∙L-1Target DNA。所以当体系内存在多种核酸甚至整个病毒碎片时,依然可以对其中含有的微量目标核酸进行检测,即对获得的咽拭子或鼻拭子采集液进行快速的病毒破坏后,GCDs-Bea与采集液进行混合后,依然能对目标核酸进行灵敏地检测,避免了核酸提纯这一工序,节省了检测时间。例如现有的核酸检测需要在采集样本后,在实验室进行30 min的紫外灭菌,再进行一系列地核酸提纯、扩增、检测(2–4 h),所以GCDs-Bea核酸检测可能为之后的检测提供新的便利。
4 结论
本研究成功合成了一种基于胍基修饰的荧光碳点(GCDs),连接分子信标(Beacon)后,可以在5 min内识别出目标核酸序列,并可检测到体系内0.005 fmol∙L-1(约300 copys∙mL-1)的核酸序列,不需要进行核酸扩增过程,并且可以在混合体系内识别出目标核酸序列,不需要进行核酸提取过程。具体过程主要为,当GCDs与Beacon结合后,GCDs与Beacon所携带的ROX荧光基团发生荧光共振能量转移,GCDs荧光被淬灭,GCDs-Bea荧光减弱,但当GCDs-Bea遇到与Beacon互补配对的目标核酸序列(Target DNA)时,Beacon与GCDs分离,GCDs荧光恢复。实验结果表明相比于目前的RT-qPCR检测方案,该方法可以缩短检测时间,提高检测效率。GCDs-Bea可以通过改变Beacon的核酸序列的方式,来达到对其他病毒或疾病的核酸检测的结果。
猜你喜欢
中华诗词(2022年9期)2022-07-29
中国慈善家(2022年3期)2022-06-14
快乐语文(2021年34期)2022-01-18
昆明医科大学学报(2021年8期)2021-08-13
中国(俄文)(2020年8期)2020-11-23
中央民族大学学报(自然科学版)(2018年3期)2018-11-09
中国医疗保险(2017年5期)2017-05-17
三峡大学学报(自然科学版)(2016年6期)2016-04-16
中国康复理论与实践(2015年10期)2015-12-24
现代电生理学杂志(2015年1期)2015-07-18
- 物理化学学报的其它文章
- Recent Advances in Self-Supported Transition-Metal-Based Electrocatalysts for Seawater Oxidation
- Performance Improvement and Antibacterial Mechanism of BiOI/ZnO Nanocomposites as Antibacterial Agent under Visible Light
- Introducing Novel,Multiple Cd Coordination Modes into Gold Nanoclusters by Combined Doping for Enhancing Electrocatalytic Performance
- 电催化二氧化碳还原催化剂、电解液、反应器和隔膜研究进展
- 利用多氟丙烯酸酯添加剂提升准二维钙钛矿发光二极管性能
- 电子自旋效应在电催化剂中的作用