
二维半导体材料的高通量计算及数据管理平台的设计与实现
2024-01-22 01:11刘小久何远德
西南民族大学学报(自然科学版) 2023年6期
刘小久,何远德
( 西南民族大学计算中心,四川 成都 610041)
随着信息技术的不断进步,计算材料学经过几十年的发展,已经从最初的对已知材料电子结构和基本特性的近似计算发展到能对材料新结构、组成进行准确预测,并发现材料未知的构效关系等.近年来我国对材料基因组计划[1]也有了自己的规划,将材料计算列为与制备和表征同等重要的地位.该计划已经从以前的对已有材料的物性解释与实验对照,发展到指导实验研究与材料合成方向的作用.要达到这样的目的,材料的高通量计算将是最关键的手段.材料性能的高通量计算[2],已经在新材料的发现中展示了巨大的作用.同时,我们看到在高通量计算的基础上,利用大数据相关技术[3],特别是机器学习[4-6]的方法、理论,为材料性能快速预测提供了新的途径和方法.材料数据科学[7]以及其相关的材料信息学[8-9]正在发展成为一门新的学科,助力材料相关研究,为新材料的研发提供全新、快捷、高效的手段.
材料的高通量计算是材料信息学的重要手段和基础,它包含三个要素[10]:材料物性的可算性、数据输入和输出的自动化以及计算任务的并行化.所谓物性的可算性,即要求材料的相关性质能够通过计算可靠地获得,它可以是能够直接计算的物理性质或者可以通过计算的物理量直接决定的特性.前者如材料的基态特性:晶体晶格常数、弹性常数以及磁化强度等等[11],这些物理量可以方便地通过基于电子密度泛函的理论计算获得;后者如拓扑绝缘体的判断,可以通过材料在相对论情况下和非相对论情况下的能带比较得到[12].自动化和并行化,是为了解决大量的计算任务提出的,只有实现了这两点,才能在有限的时间内对材料进行快速计算和筛选.比如,受电声耦合机制所决定的材料的电阻率,原则上可以通过计算的方式进行估计[13],但在目前阶段,该计算仍然需要大量的计算资源,因而无法快速地进行海量材料的计算.鉴于以上三点,目前通过高通量计算建立的数据库,主要包括合金的力学性能、化学特性[14]、Heusler化合物的输运特性[15-16],材料光催化等方面[17].除了进行第一性原理计算外,某些数据库也提供了多尺度的材料仿真和设计,实现了从材料微观到宏观性能的自动计算预测[18-19].
在实现高通量计算过程中,有必要对材料的元数据进行规范化,以便不同代码间实现数据的共享,实现计算代码的异质集成,以及材料的跨尺度计算.欧洲的NOMAD在材料电子结构的元数据标准化方面提供了一个参考[20].美国的Materials Commons[21]提供了结构材料数据的共享规范,美国国家标准局也在材料基因工程的框架下构建了材料信息学平台及相关规范(MDCS)[22].我国以北京科技大学为牵头单位也建设了材料数据库及公共服务平台[23].
为实现材料的高通量计算以及不同尺度的材料计算设计集成,需要开发相应的计算平台,以实现对数据及计算过程的自动高效管理.MIT的Ceder小组建议了一种支持高通量计算的计算基础平台以及元数据设计,并在Materials Projects平台中给出了应用的示范[24].类似的平台如美国的AFLOW[25-26],Granda MI等系统.中国科学院信息中心开发了材料计算平台Matcloud[27],主要用于二元合金的高通量计算.Material explore公司在其材料集成计算软件MedeA中也开发了相应的高通量计算模块MedeA-HT[28].以上计算平台的第一性原理计算引擎均是商业化代码VASP[29].王果等人对材料数据高性能计算数据库分配策略[30]做了相应研究,该研究着手于优化高通量计算过程中的数据库分配方案.PWscf作为一款开源的基于密度泛函理论的材料电子结构计算软件,由于其功能全面、高效可靠的特点,得到了广泛的应用,然而,基于其开发的集成计算环境以及高通量计算接口却很少,目前仅仅在AFLOW中实现了其集成化.由于开源代码的灵活性以及商业上的廉价,利用开源代码的计算平台将具有更大的市场优势及开发价值.
因而,我们基于PWscf作为计算引擎,开发了实现高通量计算的自动计算平台.在该平台中,实现了计算输入的准备、计算任务的调度管理以及数据信息的提取与分析等功能.并以二维半导体材料作为应用示范,通过自动计算,获得优化后的原子位置、生成的材料电子能带、态密度,自动计算导带低和价带顶的电子和空穴在不同方向的有效质量.同时,我们对材料的相关性能进行了统计分析,为材料构效关系的建立奠定了基础.
1 系统设计
本系统为高通量的二维材料原子位置优化、电子性能计算数据管理系统.具有可视化交互式材料原子位置录入、晶体结构显示、电子结构显示以及材料关键输运参数.如带隙大小及类型,电子和空穴有效质量自动地计算功能.针对高通量的材料计算及新材料初始结构产生,我们采用原子的组合替换方法.系统能够实现计算任务的自动生成和管理,计算过程的监控以及计算结果的提取、分析和收集入库的功能.
在总体设计上以数据库为核心,包括如下三大功能部分:材料基本信息数据库,计算节点控制作业调度管理,以及输入输出信息解析.其主要的构件及信息/数据流如下图1所示,图中箭头的方向表明数据的流向,横线定义了平台界面、软件和硬件.在该设计中,计算服务器和存储服务器由相互独立的硬件承担.计算代码和数据库条目之间的信息交换通过文件进行:由专门的脚本分析不同计算代码的输入输出文件,并利用关键字检索的方法,形成数据库条目.数据库作为数据交换的枢纽,不同代码的数据读取和存放均通过与数据库中间件的API交互完成.我们采用浏览器与数据库的交互,实现相关数据的增、删、改、查的工作以及数据的统计分析及后续处理.

图1 系统的总体架构示意图
1.1 数据库的设计
根据材料基本属性的构成,我们选择关系数据库MySQL作为材料的关键信息存储平台.主要基于以下三点考虑:首先从材料的数据级别上来看本系统属于百万级数据量,但元数据构成复杂;其次材料计算周期长,数据的成本高昂,数据需要考虑容灾备份;最后系统能够方便进行数据建模和迁移,具备未来和Hadoop大数据平台对接的基础.
根据材料基因数据的系统相关性,建立基础数据表general_data,以此为基础建立空穴、电子、带隙、赝势文件(UPF文件)等数据信息表等,见下图2.整个系统主要完成两个核心流程:
1)自动计算流程
“计算任务运维中心”基于输入条件生成批量计算脚本,“计算调度中心”收到计算任务之后分发到计算集群,由集群管理系统(PBS)完成计算任务指派和计算资源分配,并调用PWscf的相关计算模块进行计算;计算调度中心实时跟踪计算任务状态,并回报运维中心,在浏览器页面显示.
2)结果分析流程
在PBS脚本中,实现计算完成之后,回调到计算结果分析中心,对计算结果进行解读,解析计算输出文件.该流程主要获得相关体系不同k点的能量本征值,带隙以及本征能级的占据数等信息,并由计算调度中心将解析结果传递给计算运维中心,回写到数据库中.
1.2 输入/输出信息的实现
本系统涉及数据查看、查找、筛选、数据统计、材料母体结构输入、原子组合替换、原子位置优化以及材料电子结构及物性计算等过程.
为实现跨软件的计算平台,在准备计算任务的数据表示中,我们采用XML结构,将计算控制信息用伪代码表示,在生成具体计算任务时,再映射为相应的计算代码输入文件.在对输出信息提取时,我们直接读取分析PWscf所生成结果中的XML文件信息.在该文件中,包含了所有的计算输入和输出信息,包括晶格常数、原子坐标、晶体对称性等结构信息以及不同k点的本征值、Fermi能级及相应本征态的占据数等电子结构信息.它具有格式统一的特性,以及不同版本间数据结构差异小等特征,便于相关代码的编写及重用.
为实现交互式原子输入及更新内容,我们采用JMOL提供的晶体显示插件显示晶体结构.在原子位置信息表示中,我们采用较通用的POSCAR文件格式.该插件提供了对显示画面的旋转,缩放,长度和角度测量等功能,便于直观检查输入的正确性.其显示效果如图3所示.
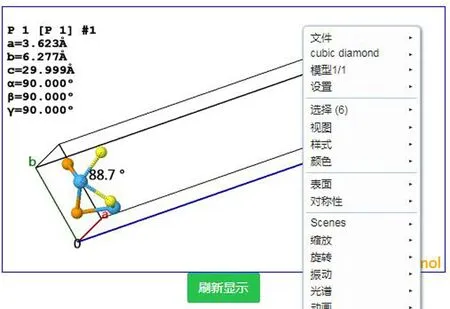
图3 基于JMOL的晶体结构显示和交互界面
1.3 计算控制
为实现高通量的计算并能够比较不同计算的结果,需要对计算精度和能带结构的输出信息做统一控制.对于PWscf,其主要的精度控制是通过控制平面波的截断动能,以及不可约布里渊区(IBZ)划分密度,即k点的间距决定.我们主要采用控制k点间距的方法,将缺省的k点间距控制在0.2 Å-1,这样的缺省值相当于在正空间的边长为3 nm立方体原胞,可以满足通常二维材料在厚度方向相互作用可以忽略的间距要求.另外,本平台同时提供自由的脚本编辑和修改功能,能够方便地根据需要设定相关的计算控制参数.
1.4 材料的组合计算
在材料的设计和计算中,材料的组合计算是产生海量不同材料的有效方法,它在新材料的发现中有重要的作用.通过计算机自动产生不同位置的元素替代,并结合第一性原理的计算,可以快速寻找到合适的材料.在二维材料中,我们也利用该方法进行新材料的产生.与通常的合金或化合物材料不同,在二维材料的生长过程中,如利用原子层沉积工艺(ALD)[31],我们能够控制新加入的原子种类,实现原子按层生长,在第三维进行材料原子剪裁.因而,在我们系统设计中,考虑到与实验的结合,采用分组的方法,实现原子替换:即将原子按原子层分组,相同分组的原子作为同一分组,同时替换.这样减少了替换的组合数,实现高效快速的计算,同时能够为实验的材料合成提供更直接相关参考.
1.5 半导体的特性参数
在本数据库中,计算并存储特了定方向的有效质量m[ijk],其中[ijk]为晶向指数,其缺省值为[100]、[010]及[001]三个正交的方向.态密度有效质量
(1)
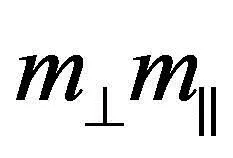
(2)
2 测试效果
二维半导体是目前研究的热点,目前,自然存在的已经发现及合成的材料仅仅几十种[32].我们可以通过不同元素的合理替代,形成新的二维材料,同时,由于不同材料在z方向弱的相互作用,这为我们利用不同的二维材料构成异质结提供了基础,因而可以利用组合的方法形成所谓的van der Waals异质结材料[33-35],为材料的特性调控中增加了新的手段和方法.利用材料组合的自动计算技术我们能迅速对材料的基本性能进行计算,并对性能变化的规律和关联性进行分析.
本数据库中我们对总共两千多种可能的单层二维材料及几类典型的van der Waals异质结二维材料进行了计算.这些单层的二维材料结构分别为MX2型, MX3型,K-C6型以及MXene,异质结材料由1H-MX2组合构成.
在本文中,我们以典型的MX2型材料为例,分析其计算获得的信息及相关统计结果.
MX2型的二硫过渡金属化合物是目前研究的热点材料,其结构如图4(a)所示:在两层硫族元素(X)中间,是过渡元素(M),根据结构是否具有中心反演对称,可以分为1H型和1T型.在平面内,MX2型二维材料的晶格为正六边形.为获得在晶格参数优化过程中的稳定性,我们采用正交的超原胞晶格.在平面内,该四边形超原胞与原来的六边形原胞的变换对应关系如图4(b)所示.
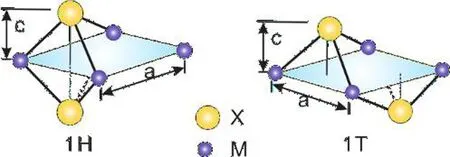
(a)结构示意图
在组合替代时,上下两层的硫族原子同时做不同的元素替换,同时过渡金属也在3d和4d元素中变化,形成X1-M-X2的化合物,这样可以获得金属到半导体(包括直接带隙和间接带隙半导体)不同带隙人工设计的新材料.并且对这些不同材料进行结构优化:包括平面内xy方向晶格常数的优化以及原子位置在z方向的弛豫以获得新材料的晶格常数,并更新数据库中用弛豫后的结构获得的电子结构、态密度、电子和空穴有效质量以及带隙等信息.这样我们可以得到材料的性能的统计信息如下:
1)物性关联
同一结构化合物的电子和空穴有效质量分布见图5(a).由图可见,有效质量具有关于45度线的对称性.从而我们可以获得电子和空穴有效质量类似的材料.同时有效质量分布具有明显的聚类特征:分布集中在图片的左下角,意味着在二维半导体中,大多数材料的电子和空穴有效质量均很小,为性能材料的选择提供了一定的空间.因为对于二维材料,粒子态密度正比于有效质量的平方根,因而,说明该体系的粒子态密度较低,这是sp电子主导的态密度的重要特征.
(a)电子和空穴平均有效质量
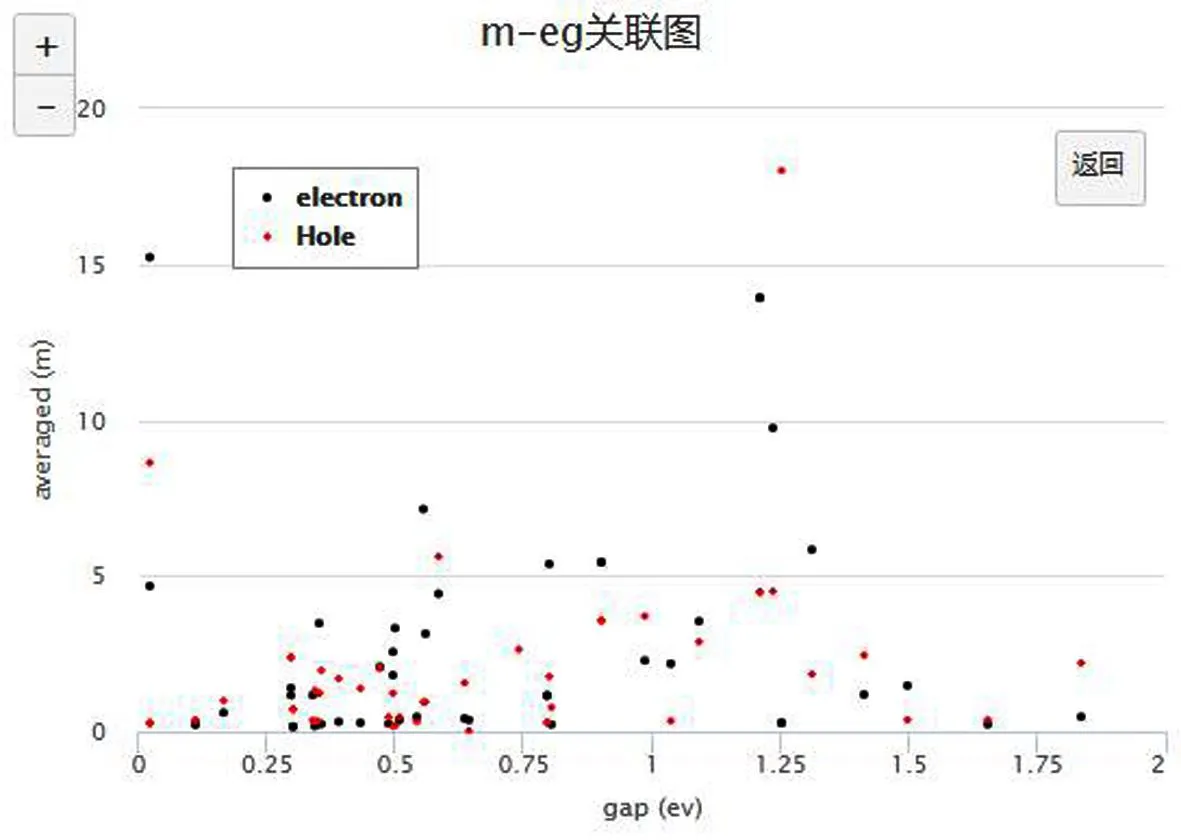
所计算的二维材料,其电子的有效质量和带隙关系的散点图如图5(b)所示.从图中可以看到,MX2型二维材料有效质量绝大部分集中在5 me以下.同时,二维材料的带隙最高可以到1.8 eV,在红光和红外光范围内,因而该类材料可能成为红外探测材料的很好候选.同时空穴有效质量普遍高于电子的有效质量,这也是通常半导体材料的共性.
2)特性统计分布
通过我们的计算,对于MX2型材料,其在[100]方向的空穴和电子的有效质量分布如图6(a)所示.从图中可以看出,材料的有效质量分布随往小的有效质量聚集,随着有效质量的增加呈明显的衰减.这个分布规律预示着我们可以在多数二维材料中获得较高的高载流子迁移率,因为在类似的材料中,其电声耦合的相互作用所形成的形变势变化在相同量级[36-37],因而,有效质量决定了材料的载流子迁移率.
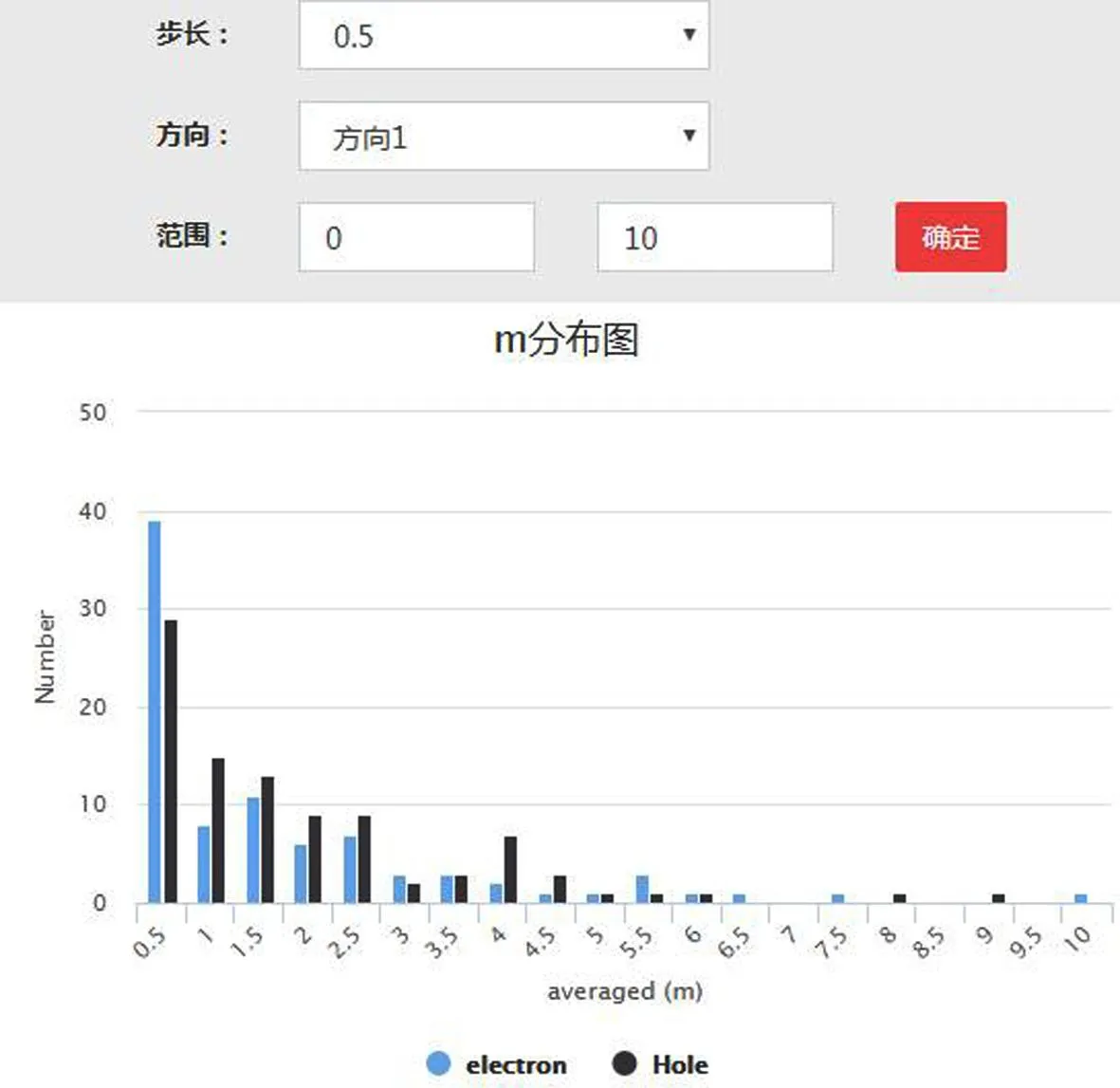
(a)电子和空穴的有效质量分布
对于其带隙值的分布也可以做类似的统计信息,如图6(b).可以看到带隙大小为0.3~0.4 eV时具有最可几分布.我们知道,在共价化合物半导体中,其带隙通常由原子的成键轨道和反成键轨道的能量差决定,而这个能量差由阴离子间相互作用决定,这种相互作用对于同族的原子相差不大,因而带隙在某一值附近获得最可几分布.
3 结论
文章介绍了本团队开发的以PWscf为计算引擎的二维半导体材料高通量计算平台的设计、实现及测试过程.该平台能够实现对计算任务的管理和调度、材料的组合计算、结构优化、基本输运性能计算,能够以友好的图形交互界面完成材料结构的输入与查看.通过在二维材料中的应用,展示了其任务管理和数据管理的功能.经过简单的数据统计,能够发现一些材料性能的变化规律,随着数据量的不断积累,有望通过数据挖掘技术、机器学习等,获得半导体关键性能的构效关系.随着材料信息学的发展以及材料大数据挖掘技术的发展和应用,平台功能的进一步完善和补充,必将为新材料的研发做出应有的贡献.
猜你喜欢
国际太空(2023年1期)2023-02-27
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
车用发动机(2021年5期)2021-10-31
电源技术(2021年7期)2021-07-29
透析与人工器官(2020年1期)2020-11-16
铁道通信信号(2019年8期)2019-10-10
赤峰学院学报·自然科学版(2018年7期)2018-08-11
中国发展观察(2017年8期)2017-04-26
