
DES磁性分子印迹材料的制备及其吸附分离性能
2024-01-13 11:23袁新华孟付良陈泽宇徐春雨费宏伟
江苏大学学报(自然科学版) 2024年1期
袁新华, 刘 帅, 孟付良, 陈泽宇, 覃 远, 徐春雨, 费宏伟
(1. 江苏大学 材料科学与工程学院, 江苏 镇江 212013; 2. 杭摩新材料集团股份有限公司, 浙江 湖州 313310; 3. 常州浩达科技股份有限公司, 江苏 常州 213133)
雌激素是指一类可影响动植物生命系统的类固醇激素.它们结构相似,进入人体后,可作用于内分泌系统、视觉系统、代谢系统和生殖系统等,干扰内分泌激素的合成和代谢,影响身体功能,破坏人体内环境的协调和稳定.己烯雌酚(DES)是一种人工合成的雌激素物质,被用于畜牧养殖业动物疾病治疗中.动物使用后,可使动物体内蛋白质同化,促进动物生长,因而被一些厂家滥用.研究[1-2]证明,DES具有致畸性和致癌性.2019年12月27日,DES被我国列入食品动物中禁止使用的药品.对样品直接进行DES检测时,干扰物会影响检测结果可靠性,因此检测前需进行前处理.
分子印迹技术(molecular imprinting technology,MIT)是模拟抗体-抗原或酶-底物之间的相互作用,对印迹分子进行专一性识别的技术[3],就如同钥匙与锁之间的对应关系.表面分子印迹技术是为了增加模板分子的识别位点、提高传质速度而发展起来的一种新的分子印迹技术.该技术是以固体材料为载体,在其表面进行聚合反应[4].聚合得到的识别位点大多位于聚合物表面,吸附与解吸附比较容易,同时可提高分子印迹聚合物对目标分子的可接近性.制备表面分子印迹聚合物方法有化学接枝法[5]、牺牲硅胶法[6]等,常用载体有二氧化硅、碳纳米管等[7].化学接枝法是目前应用比较广泛的一种表面印迹技术,即在改性后固体材料表面进行聚合反应,得到分子印迹材料.目前,将表面分子印迹技术应用于有害物吸附方面的研究取得了较大进展[8-10],但对人工合成雌激素DES的研究报道还比较少.
为此,本课题组采用表面分子印迹技术,将磁性材料(Fe3O4)作为载体,以DES为印迹分子,制备DES磁性分子印迹聚合物(MMIP),另外还制备DES磁性非分子印迹聚合物(MNIP),采用透射电镜、振动样品磁强计和吸附试验等方法进行对比,旨在对DES磁性分子印迹聚合物的吸附分离性能进行探究.
1 试验部分
1.1 试剂与仪器设备
六水合三氯化铁(FeCl3·6H2O)、无水乙酸钠(NaAc)、聚乙二醇2000(PEG2000)、乙二醇(EG)、正硅酸四乙酯(TEOS)、浓氨水、无水乙醇、甲基丙烯酰氧基丙基三甲氧基硅烷(MPS)、甲基丙烯酸(MAA)、己烯雌酚、甲苯、乙腈、甲醇和乙酸等试剂购自国药集团.雌二醇(E2)、双酚A(BPA)、偶氮二异丁腈(AIBN)和乙二醇二甲基丙烯酸酯(EGDMA)等试剂购自上海麦克林生化科技有限公司.所有试剂皆为分析纯(AR).
仪器和设备主要包括JEM-2100透射电子显微镜(日本电子株式会社)、Nicolet MX-1E傅立叶红外变换光谱仪(赛默飞有限公司)、UV-2800紫外可见分光光度计(日本岛津公司)、NOVA3000e比表面积与孔隙率分析仪(美国康塔公司)和Squid-VSM振动样品磁强计(美国Quantum Design公司).
1.2 MMIP和MNIP的合成
1.2.1Fe3O4纳米粒子的合成
利用溶剂热法制备Fe3O4纳米粒子.将8.7 g的FeCl3·6H2O加入175 mL的乙二醇溶液中,超声溶解后再加入5.1 g聚乙二醇2000和19.1 g的无水乙酸钠,在50 ℃下进行磁力搅拌,使其完全溶解,并混合均匀.将上述溶液倒入聚四氟乙烯反应釜中,把反应釜放入不锈钢外套中,然后放入烘箱,升温至200 ℃,反应8 h.所得产物用乙醇洗涤后,置于50 ℃烘箱中干燥12 h,制得Fe3O4纳米粒子.
1.2.2纳米Fe3O4表面改性
将200 mL乙醇和50 mL水加入三口烧瓶中,再加入0.3 g的Fe3O4纳米粒子,超声分散30 min,以400 r/min的转速进行机械搅拌.将4 mL质量分数为28%的氨水加入该三口烧瓶,搅拌5 min,并混合均匀.同时将5 mL的TEOS溶液缓慢滴加到烧瓶中,滴加结束后,室温下继续搅拌6 h.产物用磁铁收集,再用乙醇和蒸馏水洗涤,得到SiO2-Fe3O4纳米粒子,将其放于烘箱中干燥10 h,待用.
1.2.3SiO2-Fe3O4纳米粒子的表面改性
取120 mL甲苯加入三口烧瓶中,再加入0.3 g的SiO2-Fe3O4纳米粒子.超声分散均匀后,进行搅拌,同时缓慢滴加9 mL硅烷偶联剂MPS.滴加结束后,将烧瓶置于65 ℃水浴锅中,机械搅拌反应24 h,搅拌速率300 r/min,产物用磁铁收集,用乙醇和蒸馏水洗涤后,置于真空干燥箱中干燥12 h,得到MPS-SiO2-Fe3O4纳米粒子.
1.2.4MMIP与MNIP的合成
在100 mL三口烧瓶中加入65 mL乙腈、100 mg的DES和128 μL的MAA,混合均匀后密封,置于室温黑暗环境中,预组装12.0 h.预组装过程中,模板分子与功能单体的功能基团通过分子间的相互作用,形成具有多重作用位点的配合物,聚合后这种作用保存下来,更好地形成特异性空腔.再加入200 mg的MPS-SiO2-Fe3O4纳米粒子载体.超声分散均匀后,在搅拌的同时,加入60 mg的AIBN和1.478 mL的EGDMA溶液.然后将烧瓶置于水浴锅中,升温到65 ℃,机械搅拌下反应24.0 h,搅拌速率300 r/min.产物用磁铁收集后,分散在50 mL甲醇-乙酸洗脱液里,每隔0.5 h更换一次洗脱液,直至固体纳米材料上的DES洗脱干净.最后用乙醇和水洗涤固体纳米材料上的乙酸,得到MMIP,将MMIP置于50 ℃烘箱中干燥12.0 h待用.
DES磁性非分子印迹聚合物(MNIP)制备过程和MMIP一样,区别是制备中不加印迹分子DES.
1.3 MMIP和MNIP的吸附试验
1.3.1吸光度对质量浓度的线性拟合
配制质量浓度分别为2.5、5.0、10.0、20.0、25.0、50.0和60.0 mg/L的DES乙醇溶液.利用紫外可见分光光度计测定各质量浓度下DES乙醇溶液的紫外吸收光谱.根据最大吸收波长确定特征吸收波长,以溶液质量浓度为横坐标,以特征波长处的吸光度为纵坐标,绘制吸光度-质量浓度曲线,进行线性拟合,得到线性方程.
1.3.2MMIP和MNIP的静态吸附试验
配制质量浓度分别为25、50、100、150、200和300 mg/L的DES乙醇溶液,各取10 mL溶液分别加入6个离心管中.再各取20 mg的MMIP和MNIP分别加入上述离心管.将离心管置于恒温震荡箱中,25 ℃下振荡12 h.吸附平衡后,使用磁铁将吸附材料与溶液快速分离,并取适量上清液进行稀释.因为溶液的初始浓度已知,添加一定量溶剂就可以将其质量浓度稀释到吸光度-质量浓度线性方程要求的质量浓度线性范围内.将稀释后的溶液置于比色皿中,测定溶液在DES乙醇溶液紫外吸收光谱的特征吸收波长242 nm处的紫外吸光度,并根据紫外吸光度DES溶液质量浓度方程计算平衡质量浓度.采用下式计算平衡吸附量:
(1)
式中:Qe为平衡吸附量,mg/g;ρ0为DES溶液初始质量浓度,mg/L;ρe为DES溶液平衡质量浓度,mg/L;m为吸附材料质量,mg;V为吸附溶液体积,mL.
1.3.3MMIP和MNIP的动态吸附试验
分别取10 mL质量浓度为200 mg/L的DES溶液加入12个离心管中,平均分成两组,将20 mg的MMIP和MNIP分别加入6个离心管中.将离心管置于恒温震荡箱中,每隔10、20、40、60、100和200 min从每组中取出1个离心管.用磁铁分离、澄清浑浊溶液,取适量上清液,将其浓度稀释到吸光度-质量浓度线性方程要求的浓度线性范围内.测定溶液在DES乙醇溶液的特征吸收波长242 nm处紫外吸光度,通过DES溶液吸光度-质量浓度方程计算溶液质量浓度,采用式(1)计算其动态吸附量,此时ρe为动态吸附后的质量浓度,得到的就是动态吸附量.
1.3.4MMIP和MNIP的选择性吸附试验
各取20 mg的MMIP和MNIP,分别加入10 mL的DES、E2和BPA初始质量浓度均为200 mg/L的混合溶液中,25 ℃条件下置于震荡箱中振荡12 h.振荡结束后,通过磁铁吸附分离材料与溶液.取适量上清液,将其稀释到吸光度-质量浓度线性方程要求的浓度线性范围.测定溶液在DES乙醇溶液紫外吸收光谱的特征吸收波长242 nm处紫外吸光度,由式(1)计算MMIP和MNIP对不同吸附质的平衡吸附量.
1.3.5MMIP和MNIP的再生循环试验
在两个分别盛有10 mL初始质量浓度为200 mg/L的DES溶液烧瓶中,各加入20 mg的MMIP和MNIP进行震荡吸附.吸附3 h达到平衡后,使用磁铁将印迹材料与溶液进行分离,并取适量上清液进行稀释,再用紫外分光光度计测定吸光度,计算DES溶液平衡质量浓度.用V(甲醇)/V(乙酸)=9 ∶1的混合溶液洗脱吸附质DES,利用磁铁分离印迹材料,然后倒掉洗脱液.该操作重复6次,检查最后洗脱液中DES的紫外吸收光谱,没有DES的特征吸收峰,可将其认定为脱附完全.最后通过乙醇洗涤来除去乙酸,并继续在DES乙醇溶液中进行吸附,得到平衡吸附量.将吸附-脱附过程重复6次,分离操作均采用磁铁进行快速分离,从而检测MMIP和MNIP的再生循环性能.
2 试验结果与分析
2.1 MMIP的合成路线
MMIP的合成路线如图1所示,其中R是表面接枝的硅烷偶联剂官能团的缩写.
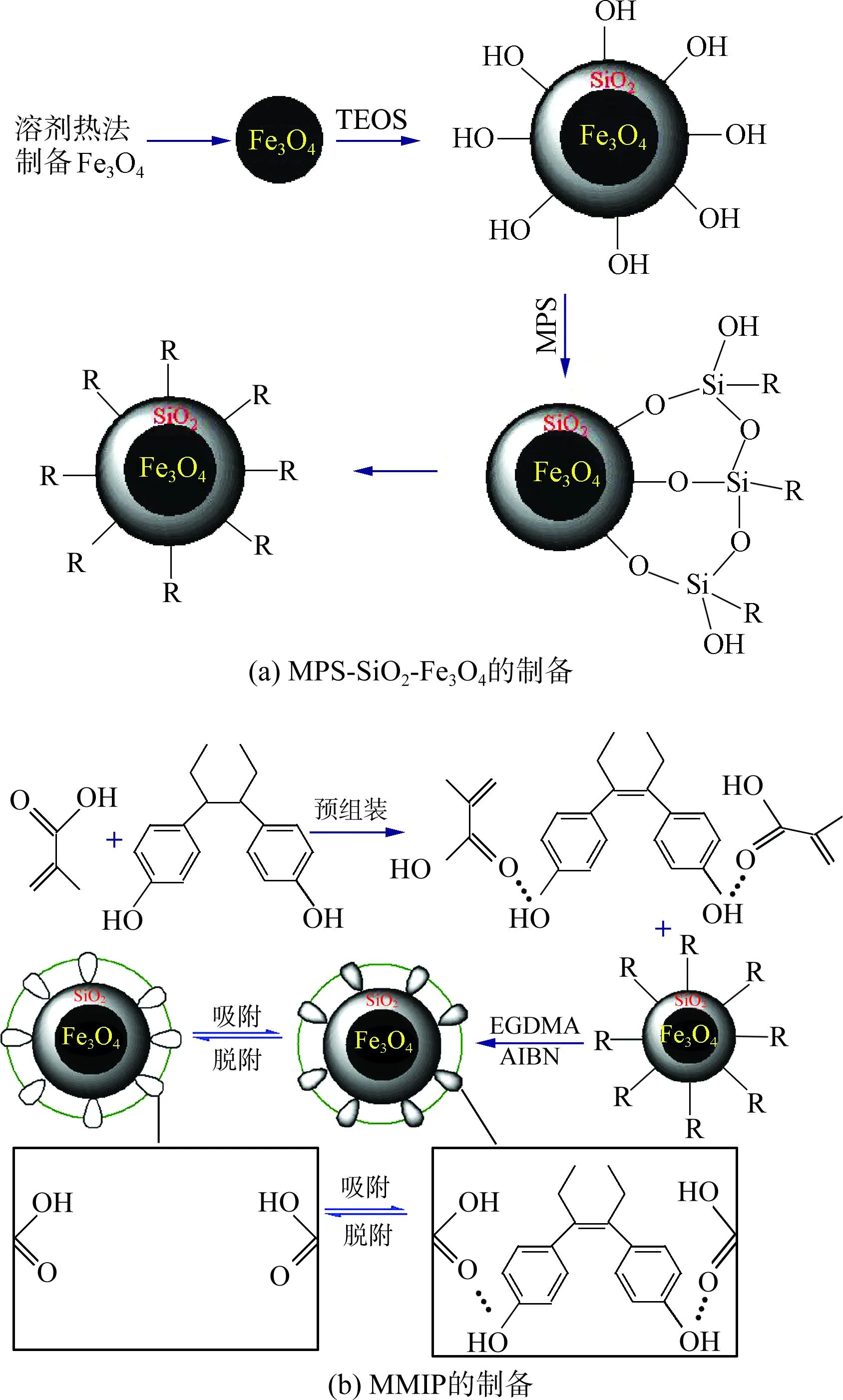
图1 MMIP合成路线示意图
首先采用溶剂热法在200 ℃下制备Fe3O4纳米粒子,然后在其表面沉积二氧化硅,再利用MPS对其进行表面接枝改性,制备分子印迹载体.将功能单体与模板分子进行自组装后,与磁性载体一起,加入交联剂、引发剂进行聚合反应.最后脱除模板分子,得到磁性分子印迹材料.由于MNIP只是没有加模板分子DES,DES的存在与否影响的是特异性腔位的形成,因而不影响MNIP合成结果.
2.2 磁强度分析及磁性分离
为了研究MMIP的磁性吸附和分离效果,用振动磁强计对样品(Fe3O4、SiO2-Fe3O4、MMIP)磁性进行了测试,同时对MMIP吸附后的磁性分离效果进行拍照.图2为Fe3O4、SiO2-Fe3O4和MMIP的磁滞回线.图3为MMIP磁性分离效果照片.
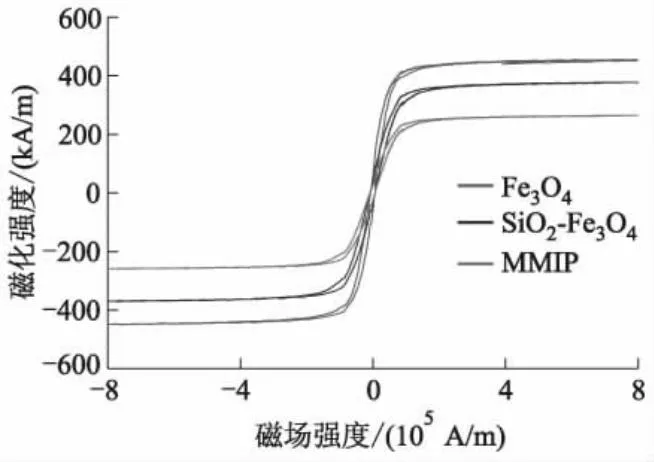
图2 Fe3O4、SiO2-Fe3O4和MMIP的磁滞回线

图3 MMIP磁性分离效果照片
由图2可知,Fe3O4、SiO2-Fe3O4和MMIP几乎无磁滞现象,矫顽力为0,具有超顺磁性,说明制备的材料利用磁铁进行磁性分离时,在磁场作用后不再有剩磁,因而可重复用于吸附和脱附操作,三者的饱和磁化强度分别为460、380和268 kA/m.这是因为SiO2在Fe3O4表面沉积,形成二氧化硅层,导致SiO2-Fe3O4磁化强度下降.功能单体与交联剂在改性后的二氧化硅层表面进行聚合反应,在表面形成分子印迹层,使得MMIP磁化强度再次降低.
由图3可知:没有外加磁场时,MMIP在溶液中悬浮,呈现浑浊液体;外加磁铁后,MMIP可在17 s内与溶液实现快速分离,溶液变得澄清透明,无需对MMIP进行离心操作,即可直接将MMIP用于紫外吸收测试,表明在外加磁场下可实现MMIP与溶液的快速分离.
2.3 透射电镜谱图
Fe3O4、SiO2-Fe3O4和MMIP的透射电镜(TEM)谱图如图4所示.由图4a、b可知,Fe3O4纳米粒子分散性较好,直径约为300 nm.图4c为正硅酸乙酯水解后,在成核剂Fe3O4粒子表面沉积、生长得到的SiO2-Fe3O4纳米粒子,可明显观察到Fe3O4纳米粒子表面的二氧化硅薄层,厚度约为30 nm.由图4d可知,磁性载体包覆层十分明显,其厚度约为60 nm,MMIP印迹层厚度约为30 nm,说明成功制备出DES磁性分子印迹聚合物.

图4 透射电镜谱图
2.4 比表面积检测法(BET)分析
表1为MMIP和MNIP的比表面积和孔径数据汇总表.由表1可知,MMIP的比表面积和孔径均比MNIP大.这是因为MMIP脱除印迹分子后,遗留下的特异性空腔会被氮气占据,使MMIP比表面积增大,空腔也使材料孔径变大.而MNIP未使用模板分子,不存在特异性空腔.

表1 MMIP和MNIP的比表面积和孔径数据汇总表
2.5 红外光谱

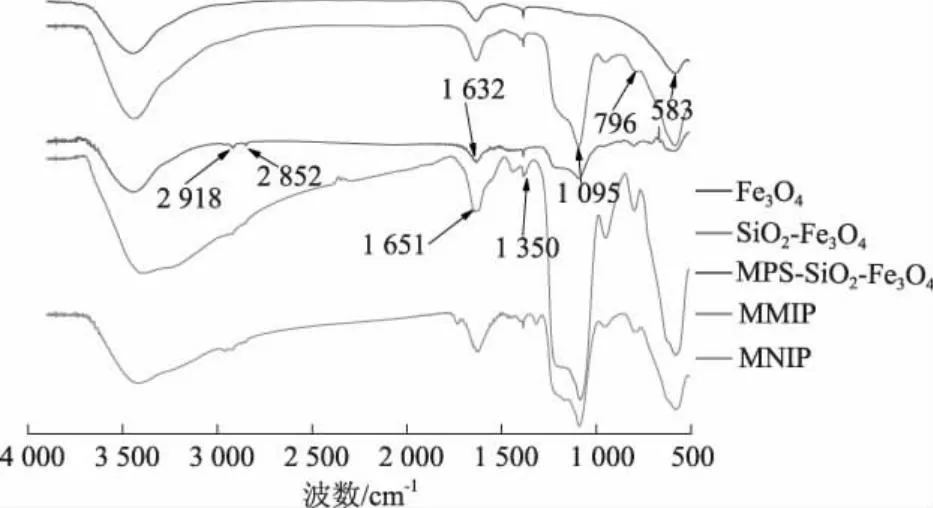
图5 红外光谱图
2.6 DES标准曲线的拟合
在一定范围内,物质的紫外吸收峰值和质量浓度呈线性关系.测定不同DES质量浓度ρ下DES乙醇溶液紫外吸收光谱,确定特征波长(见图6),并绘制吸光度与质量浓度之间相互转换的线性关系方程曲线(见图7).
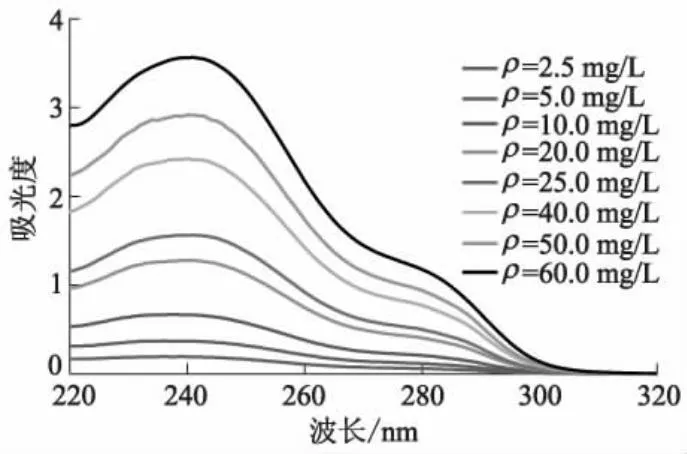
图6 不同DES质量浓度下DES乙醇溶液紫外吸收光谱图
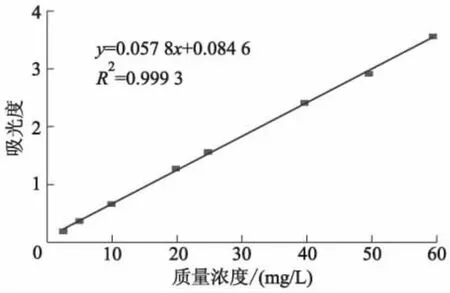
图7 吸光度与DES质量浓度线性关系拟合曲线
图6中,最大波长242 nm处的吸收峰不会随着DES质量浓度的增加而发生红移,但吸光度逐渐递增,因而DES紫外特征吸收波长为242 nm.试验中均在242 nm处测定DES紫外吸收峰.根据质量浓度和对应吸光度,拟合出的线性方程为y=0.057 8x+0.084 6,确定吸光度与质量浓度间的线性转换.
2.7 MMIP和MNIP的静态吸附
MMIP和MNIP在25 ℃下静态平衡吸附量曲线如图8所示.
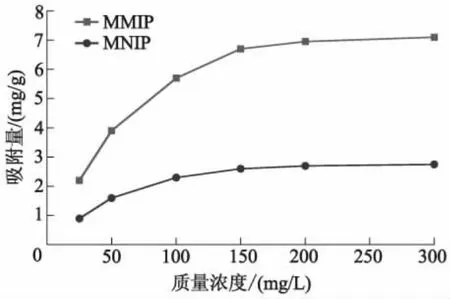
图8 MMIP和MNIP对DES的静态平衡吸附量曲线
由图8可知:随着DES初始质量浓度增加,MMIP和MNIP对DES的平衡吸附量逐渐增加;MNIP和MMIP在DES中初始质量浓度分别约为150、200 mg/L时,达到最大平衡吸附量,这是因为分子印迹材料MMIP不仅有表面吸附,还有深层吸附,而MNIP主要是表面吸附,因此较快达到吸附饱和.由图8还可知,同一初始质量浓度下,MMIP对DES的平衡吸附量始终大于MNIP.这是因为模板分子的加入,使分子印迹材料MMIP表面和内部存在着DES专一性空腔,不仅有物理吸附,同时还有化学吸附,吸附时可对DES进行专一性选择.而MNIP制备中没有加入DES,对DES的吸附主要是物理吸附,没有专一性结合位点.
2.8 MMIP和MNIP的动态吸附
图9为MMIP和MNIP对DES初始质量浓度为200 mg/L的乙醇溶液动态吸附量曲线.
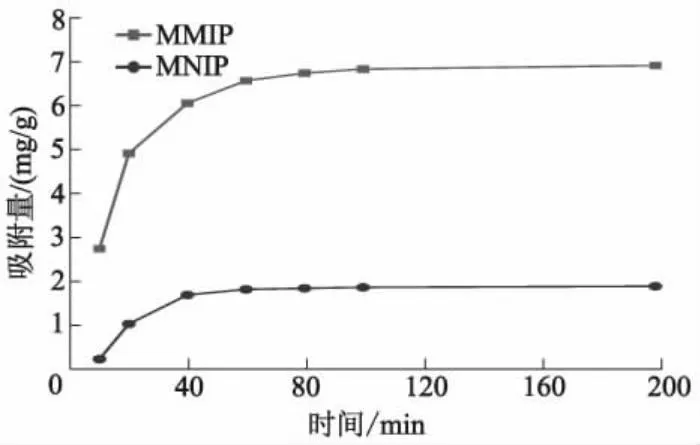
图9 MMIP和MNIP对DES乙醇溶液的动态吸附量曲线
由图9可知:一定质量浓度的DES溶液中,印迹材料和非印迹材料吸附量均随着时间t的增加而增加;t<60 min时MMIP吸附量增加较快,吸附速度变化较大,t=60 min时达到最大吸附量的90%以上,t>60 min时吸附量增加缓慢,100 min后吸附量不再增加,吸附达到平衡;t<40 min时MNIP吸附量增加较快,吸附速度也较快,t=40 min时达到最大吸附量的90%以上,t>40 min时吸附量增加缓慢,直至达到吸附平衡;吸附量增加阶段,MMIP吸附速度始终大于MNIP.开始吸附时,MMIP和MNIP通过物理吸附均可对DES进行吸附,前期吸附速度较快.由于MMIP表面具有特异性结合位点,因而可通过氢键进行化学吸附.又因为模板分子的脱除而在表面和浅表层留下印迹空腔,所以在表面吸附饱和后,MMIP会缓慢进行内部选择性吸附.MNIP没有印迹位点,只能进行物理吸附,导致MNIP达到吸附平衡的时间早于MMIP.结合位点的存在使MMIP前期吸附速度大于MNIP.MMIP平衡吸附量之所以大于MNIP,也是由于特异性印迹位点对DES的选择性吸附造成的.
2.9 MMIP和MNIP的选择性吸附
为说明磁性分子印迹材料的识别能力,在纯DES溶液以及DES与BPA、E2的混合液中进行MMIP和MNIP的选择性吸附对比.不同溶液中,二者对DES的选择性平衡吸附量对比情况见图10.

图10 MMIP和MNIP的选择性平衡吸附量对比
由图10可知:纯DES溶液中,MMIP对印迹分子DES吸附量最高,为4.89 mg/g;与纯DES溶液相比,MMIP在另外两种混合溶液中对DES的吸附量明显减少,这是BPA和E2干扰的结果,且对E2的吸附量最少;MNIP对3种物质吸附量基本相等,不具选择性,且吸附量始终小于MMIP.
表2为MMIP和MNIP对BPA、E2、DES选择性吸附量及MMIP对BPA、E2、DES的印迹因子数据汇总.
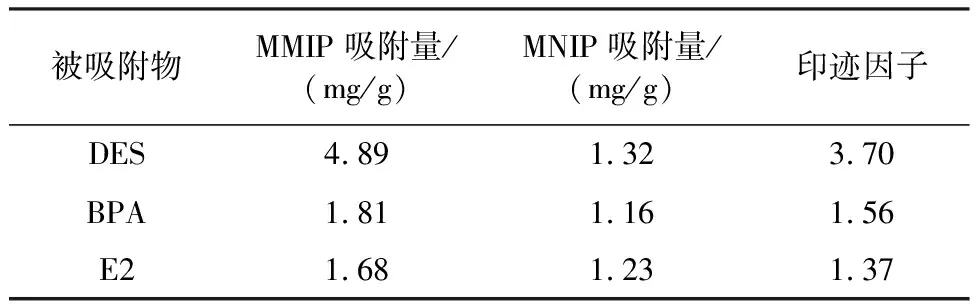
表2 MMIP和MNIP的选择性吸附量及印迹因子汇总
印迹因子越大,选择性吸附能力越强.由表2可知,MMIP对DES的印迹因子最大,可见其对DES具有较强的选择性识别能力.由于MMIP在除去模板分子后,表面留有特异性作用位点以及与模板分子相匹配的空腔,可对模板分子进行专一性识别吸附,MMIP对DES吸附效果最好.BPA与DES结构很相似,因而MMIP对其吸附量大于对E2吸附量.MNIP的吸附主要依靠物理吸附,表面没有选择性和特异性作用,因而吸附量均较小.
2.10 MMIP和MNIP的再生循环性能
再生循环性能是判断吸附材料是否具有实际应用价值的重要因素.将MMIP和MNIP分别加入10 mL的DES初始质量浓度为200 mg/L的溶液中进行吸附反应,达到吸附平衡后测定其平衡吸附量.然后洗脱模板分子,外加使用磁铁进行材料分离后,进行相同条件下的再吸附.将吸附-脱附过程重复6次,考察再生循环性能.不同再生循环次数下MMIP和MNIP的平衡吸附量对比如图11所示.

图11 MMIP和MNIP的可再生性能对比
由图11可知,随着再生循环次数增加,MMIP对DES吸附量缓慢减少,而MNIP吸附量没太大变化.MMIP吸附量减少原因可能是多次再生循环后,印迹空腔被占据或者破坏,导致结合位点减少.结果表明:MMIP再生循环6次后,吸附量仍可达到最大吸附量的90.2%.可见,MMIP具有较好的再生循环性能.
3 结 论
1) 将磁性材料和表面分子印迹技术相结合,成功制备了MMIP,其饱和磁化强度为268 kA/m,无磁滞现象,矫顽力为0 A/m,表现出超顺磁性.
2) 室温下,MMIP对DES静态最大吸附量为7.1 mg/g,在60 min时可达到最大吸附量90%以上,MMIP对DES的印迹因子为3.70,因而MMIP对DES具有特异性识别能力.
3) MMIP再生循环6次后,吸附量仍可达到最大吸附量90.2%,因而具有良好再生循环性能.
猜你喜欢
陶瓷研究(2022年3期)2022-08-19
云南画报(2021年10期)2021-11-24
生物化工(2020年6期)2021-01-07
小学生优秀作文(高年级)(2018年4期)2018-09-11
南方农业·上旬(2018年7期)2018-05-14
军事文摘·科学少年(2017年4期)2017-06-20
材料科学与工程学报(2016年2期)2017-01-15
警察技术(2015年4期)2015-02-27
中国摄影(2014年12期)2015-01-27
应用化工(2014年8期)2014-08-08
