
Brain dysfunctions and neurotoxicity induced by psychostimulants in experimental models and humans:an overview of recent findings
2024-01-10 10:13MarcelloSerraNicolaSimolaAlexiaPollackGiuliaCosta
中国神经再生研究(英文版) 2024年9期
Marcello Serra, Nicola Simola, Alexia E.Pollack, Giulia Costa,*
Abstract Preclinical and clinical studies indicate that psychostimulants, in addition to having abuse potential, may elicit brain dysfunctions and/or neurotoxic effects.Central toxicity induced by psychostimulants may pose serious health risks since the recreational use of these substances is on the rise among young people and adults.The present review provides an overview of recent research, conducted between 2018 and 2023, focusing on brain dysfunctions and neurotoxic effects elicited in experimental models and humans by amphetamine, cocaine, methamphetamine,3,4-methylenedioxymethamphetamine, methylphenidate, caffeine, and nicotine.Detailed elucidation of factors and mechanisms that underlie psychostimulant-induced brain dysfunction and neurotoxicity is crucial for understanding the acute and enduring noxious brain effects that may occur in individuals who use psychostimulants for recreational and/or therapeutic purposes.
Key Words: 3,4-methylenedioxymethamphetamine; amphetamine; caffeine; cell cultures; cocaine;methamphetamine; methylphenidate; neurotoxicity; nicotine
Introduction
Psychostimulants are a heterogeneous group of substances historically classified as “direct or indirect sympathomimetics” or “nonsympathomimetics”, according to their ability to mimic the action of the catecholamines dopamine, epinephrine, and norepinephrine (Koob et al.,2020; Additional Table 1).Sympathomimetic psychostimulants mimic the peripheral actions of catecholamines in the autonomic system by directly or indirectly activating their receptors, whereas non-sympathomimetic psychostimulants act on several neurotransmitter systems using different mechanisms of action (Koob et al., 2020).
Although indirect sympathomimetic and non-sympathomimetic psychostimulants have different mechanisms of action, as described below, both ultimately increase the synaptic levels of dopamine and other monoamines (e.g., norepinephrine and serotonin) (Rothman and Baumann,2003; Howell and Kimmel, 2007; Docherty and Alsufyani, 2021).This effect underlies their major central actions, which include elevation of mood,suppression of appetite, reduction of fatigue, and improvement of executive functions (Fast et al., 2021; Mckenzie et al., 2022).Elevated libido and heightened sexual pleasure and activity have also been documented in psychostimulant users, albeit often in conjunction with high-risk behaviors(Nazlı and Sevindik, 2020; Berry et al., 2022; Suyama et al., 2023).
Certain psychostimulants can improve brain function for specific pathological conditions and are currently used as therapeutic medications.For example,methylphenidate and dextroamphetamine, the dextrorotatory enantiomer of amphetamine (AMPH), can be prescribed to treat attention deficit hyperactivity disorder (ADHD) or narcolepsy due to their ability to increase attention, vigilance, and wakefulness (Cortese et al., 2018; Bassetti et al.,2021).Moreover, the Australian Therapeutic Goods Administration has recently announced that, starting July 2023, licensed psychiatrists will be able to prescribe 3,4-methylenedioxymethamphetamine (MDMA) to treat post-traumatic stress disorder, due to its empathogenic effects (Australian Government, Department of Health and Aged Care, Therapeutic Goods Administration).Non-sympathomimetic psychostimulants such as caffeine and nicotine can be used as performance enhancers by certain professionals(e.g., aviation, military corps, and truck drivers) for their anti-fatigue effects(Gore et al., 2010; McLellan et al., 2019; Kagabo et al., 2020).Indeed, caffeine has been reported to elevate mood and working memory, even though some authors have postulated that these effects could be due to its reversal of caffeine withdrawal (Lin et al., 2023).Moreover, both preclinical models and human studies have demonstrated that nicotine enhances cognitive performance by improving learning, memory, and attention (Valentine and Sofuoglu, 2018).
At the same time, the ability of psychostimulants to increase the synaptic levels of dopamine also underlies their ability to generate addictive behaviors,psychotic-like symptoms, and deficits in executive functions (Volkow and Swanson, 2003).AMPH, cocaine, methamphetamine (METH), and nicotine possess high abuse liability and may induce overt addictive behaviors in users.While the existence of addictive-like behaviors has also been suggested for users of caffeine and MDMA, drug consumption behaviors in these individuals rarely meet substance abuse criteria according to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition.Psychotic-like symptoms may be observed in users of AMPH, METH, or cocaine, all of which increase extracellular levels of dopamine by acting at the dopamine transporter (DAT).For example, cocaine can elicit symptoms such as paranoia, hallucinations,violence, and aggression, even following a single administration (Gicas et al.,2022); similar effects have been observed in AMPH and METH users (Morton and Stock, 2000; Gicas et al., 2022).In contrast, users of caffeine, MDMA,or methylphenidate are less prone to exhibit psychotic-like behaviors (Patel et al., 2011; Virani et al., 2018).Nevertheless, MDMA consumption can be associated with anxiety and depressed mood, as well as deficits in working memory, attention, and verbal processing (Costa and Gołembiowska, 2022;Montgomery and Roberts, 2022).Moreover, caffeine consumption may facilitate relapse in psychiatric patients (Rizkallah et al., 2011).
The manifestation of behavioral abnormalities in psychostimulant users suggests that brain dysfunctions, and possibly neurotoxic phenomena,may occur in these individuals, especially in brain regions that regulate cognitive and emotional domains.Although the induction of neurotoxicity by psychostimulants has not conclusively been demonstrated in humans,imaging studies have reported the existence of altered brain morphology and connectivity in psychostimulant users (Boxler et al., 2018; Vuletic et al., 2018;Elbejjani et al., 2019; Bittencourt et al., 2021; Avram et al., 2022).Importantly,psychostimulant-induced alterations in brain function depend on several factors, such as the amount of drug consumed, the routes of drug intake,and the age and sex of the consumer (Moratalla et al., 2017).Indeed, it is noteworthy that prescription psychostimulants (e.g., AMPH, methylphenidate)may elicit detrimental, rather than beneficial, effects on the brain when they are taken outside of a therapeutic plan (e.g., at higher amounts and/or for longer times than prescribed).Moreover, people who use psychostimulants for recreational purposes often consume multiple psychostimulants (polydrug use), which further complicates ascribing a specific substance with the induction of definite dysfunctional/neurotoxic effects in the brain (Rudin et al., 2021).
To elucidate risk factors (e.g.age, dose, sex) and mechanisms related to psychostimulant-induced neurotoxicity, the present review provides a narrative overview of recently published findings in experimental models and humans regarding brain dysfunctions and neurotoxic effects following administration/consumption of the most used psychostimulants worldwide:the sympathomimetics AMPH, methylphenidate, cocaine, METH and MDMA,and the non-sympathomimetics caffeine and nicotine.The implications of these findings are briefly discussed in terms of risks associated with psychostimulant use.Due to its concise format, the present review focuses on brain dysfunctions and neurotoxicity induced by psychostimulants; it does not provide extensive coverage of the beneficial effects of psychostimulants on brain function.
Search Strategy
We began with an extensive search strategy to conduct a comprehensive review of the relevant literature, focusing on original research articles, metaanalyses, and review articles published in the last 5 years (2018–2023).Articles were searched by using the specific search terms “psychostimulants”AND “brain dysfunction” AND “neurotoxicity” in PubMed, to focus on articles on the brain dysfunction and neurotoxicity induced by AMPH, cocaine, METH,MDMA, methylphenidate, caffeine, or nicotine in experimental models and humans.The initial search yielded 296 articles.After reading the titles and abstracts of these articles, we included in the search strategy a series of additional keywords (e.g., the name of specific psychostimulants) focusing on the drugs’ biochemical effects and mechanisms of toxicity at the central level, rather than on the behavioral effects of specific psychostimulants.Pure behavioral studies were not included in the present review.When a limited number of relevant articles were found, we adjusted the search criteria, for example by extending the search period to the most recent articles available,in order to obtain more relevant literature.Finally, we conducted filtering and article selection in accordance with the journal’s criteria (e.g., selection of no more than three articles from the same author).Overall, the recent findings reported in this narrative review are derived from 120 articles published between 2018 and 2023.In consideration of the journal’s limitations on the number of references, we could not include all possible relevant literature.1999), bioenergetic failure (Wan et al., 1999) and increased production of reactive oxygen (ROS) and nitrogen (RNS) species (Bashkatova et al.,2004; Wan et al., 2006; Figure 1).Recent evidence in rats indicates that the different mechanisms underlying AMPH-induced neurotoxicity may occur sequentially, with energy failure preceding excitotoxicity and excitotoxicity preceding the production of free radicals (Tung et al., 2017; Additional Table 2).Moreover, a study using PC12 cells suggests that the protein phosphatase 2A (PP2A)/AKT/glycogen synthase kinase-3beta (GSK3β) pathway may be an important player in AMPH-induced apoptotic cell death (He et al., 2018;Additional Table 2).Preclinical investigations in mice, rats, and nonhuman primates have demonstrated that systemic administration of AMPH, either as a racemic mixture or dextroamphetamine, may induce marked neurotoxicity in striatal dopaminergic terminals, the severity of which varies depending on the dosing and frequency of AMPH administration.Indeed, AMPH may reduce the synthesis and levels of dopamine, as well as the density of tyrosine hydroxylase (TH) and of DAT in the caudate-putamen nucleus (CPu) (Ricaurte et al., 2005; Levi et al., 2012; Tung et al., 2017), although a progressive recovery of striatal dopaminergic functionality has been also reported with time (Melega et al., 1997).Conversely, and differently from what observed after the administration of other AMPH-related drugs, AMPH does not induce neurotoxic damage in dopaminergic neurons located in the substantia nigrapars compacta(SNc) (Gramage et al., 2010).Results from a recent preclinical investigation suggest that AMPH may induce the appearance of motor deficits before the emergence of signs of dopaminergic neurotoxicity.Indeed, Apóstol del Rosal et al.(2021) demonstrated that repeated administration of AMPH in rats induced microgliosis and astrogliosis in the dorsal CPu paired with an impairment in locomotor activity and motor coordination, but without affecting the immunoreactivity of TH-positive dopaminergic neurons and terminals in the nigrostriatal system (Apóstol del Rosal et al., 2021; Additional Table 2).In addition, AMPH may induce neuronal degeneration in nondopaminergic populations, most notably in the hippocampus, parietal cortex,and piriform cortex, as demonstrated by studies in adult rats (Bowyer et al.,2008).
Indirect Sympathomimetic Psychostimulants
Indirect sympathomimetic psychostimulants have chemical structures that resemble that of the catecholamines.Cocaine, AMPH, and the AMPH-related drugs methylphenidate, METH, and MDMA are all indirect sympathomimetics.These drugs bind to DAT and to the norepinephrine and serotonin (SERT)transporters to increase the synaptic levels of monoamines (John and Jones, 2007).The precise mechanism by which indirect sympathomimetic psychostimulants exert their central effects is not yet fully defined.It is widely believed that these drugs produce their effects by enhancing dopamine neurotransmission in the brain, and in particular by inducing DAT-mediated reverse transport and/or by blocking dopamine reuptake through DAT (Koob et al., 2020).These drugs are also called “psychomotor stimulants” because their primary biological activity, as indirect dopamine agonists, is to produce behavioral activation, which is usually expressed as arousal, alertness, and increased motor activity (Koob et al., 2020).
AMPH and AMPH-related drugs also bind to the vesicular monoamine transporter-2 (VMAT-2) (Freyberg et al., 2016; Cholanians et al., 2019;Jayanthi et al., 2021), thus inhibiting vesicular uptake of dopamine and promoting dopamine release from synaptic vesicles, thereby increasing the amount of cytosolic dopamine available for reverse transport by DAT (Freyberg et al., 2016).The enhanced synaptic availability of monoamines results in the activation of receptors for dopamine, norepinephrine, and serotonin in the mesocorticolimbic and nigrostriatal systems (Drouin et al., 2002; Salamone and Correa, 2012; Müller and Homberg, 2015).
Amphetamine
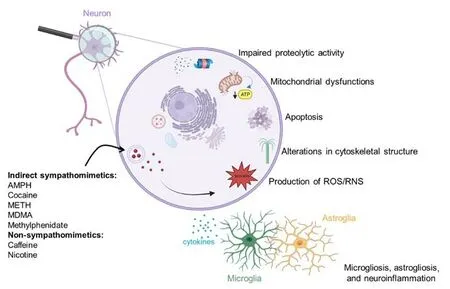
Figure 1 |Scheme of the proposed mechanisms contributing to psychostimulantinduced brain dysfunctions and neurotoxicity.Created with BioRender.com.AMPH: Amphetamine; MDMA:3,4-methylenedioxymethamphetamine; METH: methamphetamine; RNS: reactive nitrogen species; ROS: reactive oxygen species.
AMPH is a psychostimulant utilized as a racemic mixture or as dextroamphetamine for the treatment of ADHD, narcolepsy, and binge eating disorder (Cortese et al., 2018; Bassettiet al., 2021).Besides its therapeutic application, AMPH is popular as a recreational drug due to its stimulant and euphoriant effects.According to recent self-reported survey responses, it was estimated that approximately 36 million individuals had used AMPHrelated drugs (either AMPH or METH) in 2021, with a male-to-female ratio of 55:45 (UNODC, 2023).AMPH is highly consumed in Europe, where it ranks as the second most used psychostimulant drug after cocaine (UNODC,2023).While AMPH-based prescriptions are safe when used for therapeutic purposes (Weyandt et al., 2016), the recreational use of AMPH raises concerns since users could be exposed to high and potentially neurotoxic amounts of the drug.In this respect, several pathological mechanisms have been implicated in the neurotoxic effects of AMPH including, but not limited to, hyperthermia (Miller and O’Callaghan, 1995), apoptosis (Stumm et al.,Neuroimaging studies suggest that neurotoxic effects of AMPH may also occur in humans, which is consistent with evidence that individuals with a history of AMPH consumption may display deficits in memory, decisionmaking, and set-shifting (Ersche et al., 2005; Schouw et al., 2013).A recent meta-analysis evaluated the occurrence of dopaminergic dysfunction in stimulant users, by comparing 31 positron emission tomography (PET) and single-photon emission computed tomography (SPECT) studies, revealing a significant reduction in the striatal availability of dopamine D2 (D2R) and D3(D3R) receptors, as well as of DAT, in recreational AMPH users (Ashok et al.,2017).Conversely, striatal dopamine synthesis capacity, VMAT availability,and expression of dopamine D1 (D1R) receptors remained unaffected in this population (Ashok et al., 2017).Moreover, magnetic resonance imaging(MRI) investigations demonstrated that chronic AMPH users showed lower striatal activity in anticipation of reward (Schouw et al., 2013), as well as reduced cortico-striatal-thalamic and prefrontal-limbic-thalamic connectivity when acutely exposed to AMPH (Schrantee et al., 2016; Avram et al.,2022).Intriguingly, Avram and collaborators also reported that acute AMPH administration to healthy volunteers may promote auditory-sensorimotorthalamic hyperconnectivity (Avram et al., 2022; Additional Table 2), a finding that mirrors what has been previously observed in psychiatric subjects(Woodward and Heckers, 2016), and highlights the inability of the thalamus to properly filter incoming sensory signals.
Cocaine
Approximately 22 million people are estimated to have used cocaine or cocaine-containing products at least once in 2021, according to the World Drug Report (2023).These figures make cocaine the first or second most used drug of abuse worldwide, according to the country considered.The widespread use of cocaine is related to its potent psychostimulant action,resulting in euphoria, increased energy, and mental alertness (Frank et al.,1992; Hutten et al., 2018).Cocaine is generally available in two forms: as a free base (or “crack”), consumed by smoking, and as a salt, consumed by snorting or injection.
The central effects of cocaine stem from its blockade of the reuptake of monoamine neurotransmitters, causing an increase in their synaptic concentrations (Koob et al., 2020).The DAT is a major target of cocaine;increased extracellular levels of dopamine in the mesocorticolimbic system are responsible for cocaine-induced euphoria and sense of well-being, but also for its addiction potential and other central untoward effects, such as irritability and induction of psychotic-like states (Koob et al., 2020).It is noteworthy that dopamine can undergo autooxidation, and several studies have suggested that extracellular dopamine may readily autoxidize to produce ROS, which are responsible for oxidative stress (Schieber and Chandel, 2014),one of the causative factors of neurotoxicity (Rudin et al., 2021; Figure 1).Another contributing factor to the neurotoxic effects of cocaine may be neuroinflammation.Research conducted using bothin vitroandin vivomodels has demonstrated that cocaine activates microglia, leading to the release of pro-inflammatory mediators, particularly in brain regions such as the prefrontal cortex (PFC), ventral tegmental area (VTA), and nucleus accumbens (NAc) (Liao et al., 2016; Vallender et al., 2017; Mai et al., 2019;Thangaraj et al., 2020).Interestingly, evidence also indicates that cocaineinduced production of pro-inflammatory mediators may stem from pericytes,a cellular constituent implicated in the preservation and integrity of the bloodbrain barrier, as the result of impaired autophagy (Sil et al., 2019).In humans,a recent study has substantiated preclinical data by demonstrating that cocaine use disorder alters gene networks implicated in neuroinflammation in the NAc and CPu (Mews et al., 2023); however, other studies have failed to demonstrate microgliosis in cocaine users (Narendran et al., 2014).
Collectively,in vitroandin vivostudies have suggested that cocaine may induce dysfunctions and structural damage in cortical and mesencephalic regions (Camarini et al., 2017; Hirsiger et al., 2019; Bittencourt et al., 2021;Clare et al., 2021; Nicolucci et al., 2021; Tondo et al., 2021; Angarita et al.,2022).While some debate persists, these pathological alterations may be linked to cocaine’s capacity to induce mitochondrial dysfunction and increase the vulnerability of neuronal and glial cells, thereby promoting cell death via apoptosis (De Oliveira and Jardim, 2016).
Recent studies in experimental animals indicate that the ability of cocaine to induce synaptic dysfunctions depends on the age and sex of the animals.Zhu et al.(2017) reported that rats treated with cocaine during adolescence showed behavioral abnormalities at adulthood, associated with neurochemical alterations in specific brain regions (Additional Table 2).For example, compared to rats treated with vehicle during adolescence, adult rats exposed to cocaine as adolescents exhibited cognitive deficits along with increased locomotor and anxiety-like behaviors, paired with a decrease in the levels of synapse-related proteins (synapsin I and PSD-95) and in the density of synapses and dendritic spines in the medial prefrontal cortex (mPFC) (Zhu et al., 2017; Additional Table 2).The finding that cocaine administration during adolescence induced altered morphology in the rat brain at adulthood suggests that cocaine may have long-term neurotoxic effects that persist after drug exposure.On the other hand, as emphasized by recent reviews on the topic, it is noteworthy that alterations in cognitive function, which may potentially be related to neurotoxic events, can be either mitigated or exacerbated depending on the timing of cocaine use during adolescence(Kantak, 2020; Caffino et al., 2022).These data in experimental animals are relevant to humans, since it is during adolescence that the use/abuse of illicit drugs, including cocaine, typically begins, as demonstrated by several longitudinal studies (Gerra et al., 2020; Moska et al., 2021).With regard to the possible influence of sex on the neurotoxic effects of cocaine, Clare et al.(2021) investigated changes in total neuronal density measured by NeuN(a marker of mature neurons) and the concentration of neurons expressing D2R in the mPFC, dorsal CPu, VTA, and NAc in both male and female mice treated with cocaine (Clare et al., 2021; Additional Table 2).They reported that cocaine-treated female mice had higher numbers of D2R-expressing neurons in the mPFC and NAc compared to vehicle-treated female mice, and also in the dorsal CPu and VTA compared to vehicle- and cocaine-treated male mice.Moreover, cocaine-treated female mice displayed higher D2R mRNA levels in the dorsal CPu, compared to their respective vehicle-treated female controls, as well as higher D2R and proenkephalin mRNA levels in the mPFC,compared to vehicle- and cocaine-treated male mice (Clare et al., 2021;Additional Table 2).Nevertheless, cocaine-treated female mice showed a decrease in the number of NeuN-positive neurons in the dorsal CPu and VTA,compared to saline-treated female mice, and also in the mPFC, compared to saline- and cocaine-treated male mice (Clare et al., 2021; Additional Table 2).On the other hand, cocaine-treated male mice had a higher number of D2Rexpressing neurons in the dorsal CPu, NAc, and VTA, higher prodynorphin mRNA levels in the VTA, higher proenkephalin mRNA levels in the NAc and less NeuN-positive neurons in the mPFC, NAc, and VTA, compared to male controls (Clare et al., 2021; Additional Table 2).Taken together, these observations are of particular interest, since they demonstrate the existence of sex-related differences in cocaine-induced neurotoxicity and identify D2R expressing neurons as a target of this effect.In humans, imaging studies have demonstrated the existence of morphological and functional differences in specific brain regions between cocaine users and non-users.These differences, though not always significant, have been identified in several cortical regions (e.g., anterior cingulate cortex, cortical insula, lateral frontal cortex) (Hirsiger et al., 2019; Bittencourt et al., 2021),CPu (Ashok et al., 2017) and NAc (Schuch-Goi et al., 2017).Abnormalities in the brains of cocaine users have also been found in white matter microstructure, suggesting the presence of demyelination and axonal damage(King et al., 2022; Gaudreault et al., 2023).Moreover, evidence indicates that cocaine-induced synaptic dysfunctions and neurotoxicity in cocaine users may not necessarily be affected by the duration of drug use and sex of the user.An MRI study by Bittencourt and coworkers (2021) reported that, compared to healthy controls, male crack users displayed decreased cortical thickness in the left inferior temporal cortex, which was negatively correlated with the duration of crack use (Bittencourt et al., 2021; Additional Table 2).Moreover,another MRI study by Abdel Malek and et al.(2022) found no significant sexspecific differences in the morphometry of the cortical insula between male and female cocaine users (Additional Table 2).
Methamphetamine
METH is an extremely addictive psychostimulant with significant abuse liability, characterized by rapid brain penetration and quick onset of its desired effects.According to the World Drug Report (2023), METH is the second most used illicit psychostimulant worldwide after cocaine.Traditionally,METH use has been concentrated in North America, but more recently, there is increasing presence of METH in East and South-East Asia, South-Eastern Europe, as well as Australia and New Zealand (UNODC, 2023).In the United States, METH ranked as the fourth most implicated drug in overdose deaths in 2017 (Hedegaard et al., 2019).
Multiple pathological alterations follow METH administration, which likely contribute to its neurotoxic potential.Among these are hyperthermia (Masai et al., 2021), dysfunction of the proteasomal system (Meng et al., 2020),increased autophagy (Subu et al., 2020), apoptosis (Huang et al., 2019),persistent neuroinflammation (Xu et al., 2023), oxidative and nitrosative stress (Xie et al., 2018; Qiao et al., 2019), and programmed necrosis (Zhao et al., 2021; Figure 1).Moreover, recent studies indicate the disruption of the gut microbiota as a potentially novel mechanism implicated in the neurotoxic effects of METH.Indeed, METH can decrease gut microbial diversity, promote intestinal colon inflammation, and damage its barrier functions, thereby compromising the release of anti-inflammatory metabolites (especially sphingolipids and serotonin) from the intestinal microbiota into the systemic circulation (Zhang et al., 2022; Additional Table 3).
Rodents and nonhuman primates receiving either acute administration of high doses or repeated “binge” administration of METH display neurotoxicity in the nigrostriatal and, to a lesser extent, mesolimbic dopaminergic systems(Moratalla et al., 2017).In line with previous reports, recent investigations in rodents that have evaluated the impact of METH-induced dopaminergic dysfunctions on cognitive performance and drug-taking behavior have confirmed a significant reduction in TH, DAT, and VMAT-2 proteins, along with a decrease in the concentration of dopamine and its metabolites in the CPu of METH-treated mice (Masai et al., 2021; Huang et al., 2022) and rats(Schweppe et al., 2020; Jayanthi et al., 2022).Interestingly, these studies demonstrated that while METH-induced variations in monoaminergic markers are not correlated with the occurrence of cognitive deficits in mice(Schweppe et al., 2020; Additional Table 3), these neurochemical changes might be useful to predict animals’ sensitivity to the addictive effects of METH(Jayanthi et al., 2022; Additional Table 3).Moreover, these studies highlight the key role of neuroinflammation and oxidative stress in METH-mediated neurotoxicity.Indeed, either blockade of high mobility group box-1 (HMGB1),a nuclear transcriptional activator that drives inflammatory responses (Masai et al., 2021; Additional Table 3), or activation of pathways that regulate the cytoprotective responses to ROS/RNS (Huang et al., 2022; Additional Table 3), attenuated the detrimental effects of METH on the dopaminergic system.These findings hold substantial significance, especially consideringin vitroevidence that established a direct correlation between HMGB1 protein levels and D2R expression (Mao et al., 2015).The latter finding, in particular, may contribute to enhancing our understanding of the mechanisms underlying the robust protection against METH-induced dopaminergic neurotoxicity that has been observed in D2R knock-out mice (Granado et al., 2011).
Acute or binge administration of high doses of METH can also induce the degeneration of dopaminergic neurons in the SNc of mice, rats, and nonhuman primates (Harvey et al., 2000; Kousik et al., 2014; Shin et al.,2014), as well as severe deficits in motor activity and coordination in rats and mice (Jiang et al., 2014; Dang et al., 2017; Shin et al., 2019).Although a partial recovery has been reported over time (Granado et al., 2018), evidence in adult male rats indicates that METH-induced dopaminergic neurotoxicity persists for months after treatment discontinuation (Schweppe et al., 2020).
Besides affecting dopaminergic neurons, METH administration may impact brain regions that contain serotonergic (Silva et al., 2014), γ-aminobutyric acid (GABA)-ergic (Fujáková-Lipski et al., 2017), and glutamatergic neurons(Zhang et al., 2014).Recent findings in mice have shown that repeated METH administration negatively affects synapses located in the PFC and hippocampus, leading to lower synaptic density, loss of mature spines,and compromised post-synaptic structure (Ding et al., 2022; Additional Table 3).Other studies reported that METH increases the expression of neuroinflammatory, such as tumor necrosis factor-α and pro-apoptotic(e.g.caspase-3) markers in the hippocampus of rats (Shafahi et al., 2018;Additional Table 3).In addition, blood-brain barrier leakage and lowered signaling for 4-[18F]ADAM/PET, anin vivomarker of SERT availability, have been reported in the hypothalamus, thalamus, hippocampus, and PFC of METH-treated rats (Lafuente et al., 2018; Huang et al., 2019; Additional Table 3).In female cynomolgus monkeys, both acute and chronic METH treatments were found to reduce the hippocampal gray matter volume and to alter the expression of genes implicated in vesicle localization, cytoskeleton organization, synaptic transmission, and regulation of neuronal differentiation and neurogenesis (Choi et al., 2018; Additional Table 3).
In line with preclinical findings, studies in humans indicate that METH use may be associated with neurotoxic damage at the level of dopaminergic and serotonergic systems, along with neuroanatomical, neurochemical, and functional alterations in cortical and subcortical structures (Moszczynska and Callan, 2017).For example, people with a history of METH use have a 2-to-3-fold increased risk to develop Parkinson’s disease (PD) compared to the general population (Lappin et al., 2018), which supports the possible occurrence of neurotoxicity at the level of mesencephalic nigrostriatal dopaminergic neurons.
Neuroimaging studies in humans have demonstrated the presence of several structural and functional abnormalities in the brain of METH users, with either a reduction or an increase in gray matter volume (London et al., 2015).In more recent studies, the thickness of cortical gray matter was negatively associated with cumulative drug intake and drug craving in METH users (Okita et al., 2016; Additional Table 3).In addition, loss of integrity in the white matter has been reported in METH users, as measured by changes in the mean diffusivity and fractional anisotropy (FA) (Beard et al., 2019; Ghavidel et al., 2020; Ottino-González et al., 2022; Zhou et al., 2023).
Changes in biological markers of neuronal function have also been detected in METH users, such as a decreased glucose metabolism in the left insula, leftprecentral gyrus, and anterior cingulate cortex (Vuletic et al., 2018; Additional Table 3).Furthermore, a recent proton magnetic resonance spectroscopy study identified pathological features regarding levels of n-acetyl-aspartate,myo-inositol, and glutamate in the mPFC of METH users, likely reflecting a decrease in neuronal integrity and the occurrence of mitochondrial dysfunction (Wu et al., 2018; Additional Table 3).
Even though neuroimaging studies strongly suggest the existence of neurotoxic phenomena in recreational METH users, conclusive evidence is still lacking (Kish et al., 2017).Moreover, earlier studies have observed that METH-associated abnormalities in gray matter volume, DAT density, and levels of vesicular dopamine may recover after protracted abstinence from METH(Volkow et al., 2015).Nonetheless, the recovery of monoaminergic markers does not excludeper sethe possibility that METH may induce neuronal degeneration, since the increased synthesis of monoaminergic markers may occur in the neuronal population that remains following a neurotoxic insult;this adaptive mechanism may reflect an attempt to curb the loss of neuronal signaling, as previously observed in the context of neurodegenerative diseases, such as PD (Brotchie and Fitzer-Attas, 2009).
3,4-Methylenedioxymethamphetamine
MDMA, also known as “ecstasy” or “Molly”, is a ring-substituted AMPHrelated drug extremely popular due to its stimulant and euphoriant effects.According to the World Drug Report (2023), in 2021 around 20 million people aged 15–64 had used MDMA in the past year, with a male-to-female ratio of 62:38.
Several pathological mechanisms have been implicated in the neurotoxic effects of MDMA.Among them are hyperthermia (Capela et al., 2006),formation of ROS/RNS (Górska et al., 2014), alterations in cytoskeletal structure (García-Cabrerizo and García-Fuster, 2015), apoptosis (Bakhshayesh et al., 2017), mitochondrial complex inhibition associated with bioenergetic metabolic dysfunction (Taghizadeh et al., 2016) and astrogliosis (Miner et al.,2017; Figure 1).
In rats and nonhuman primates, MDMA elicits neurotoxicity mainly in serotonergic systems.Indeed, MDMA has been found to reduce striatal,limbic, and cortical levels of serotonin, to decrease the activity of tryptophan hydroxylase, the rate-limiting enzyme in serotonin biosynthesis, and to lower SERT density and availability (Biezonski and Meyer, 2010; Beaudoin-Gobert et al., 2015; Lizarraga et al., 2015; Shih et al., 2016).These noxious effects may persist long after administration of MDMA.In this respect, a recent PET/MRI follow-up investigation conducted in Formosan rock monkeys demonstrated that the neurotoxic effects of MDMA on the serotonergic system may persist even 66 months after drug discontinuation (Yeh et al., 2022; Additional Table 4).Interestingly, Yeh and coworkers also reported a region-specific selfrecovery of SERT availability in the occipital and cingulate cortex, but not in the CPu, hippocampus, thalamus, hypothalamus, midbrain, amygdala, frontal and orbitofrontal cortex (Yeh et al., 2022).
Evidence suggests that the neurotoxic effects of MDMA extend beyond the serotonergic system.Two recent studies have demonstrated that MDMA may induce neurodegeneration in the dopaminergic system of both rats(Cadoni et al., 2017) and macaques (Millot et al., 2020).Indeed, rats exposed to multiple MDMA injections during adolescence showed marked signs of dopaminergic damage at adulthood, as indicated by a reduced number of TH-positive neurons in the SNc and VTA, and decreased immunoreactivity for DAT and TH in the CPu (Cadoni et al., 2017; Additional Table 4).Moreover,Millot and coworkers (2020) reported that repeated MDMA treatment may elicit a significant reduction in DAT availability in the CPu of male macaques,along with lower SERT availability in the CPu and thalamus (Additional Table 4).The same study also found that prior exposure to MDMA aggravated the parkinsonian-like symptoms induced by the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in macaques (Millot et al.,2020; Additional Table 4).
In contrast to rats and nonhuman primates, the neurotoxic effects of MDMA in mice are more evident in the dopaminergic nigrostriatal and mesolimbic systems (Moratalla et al., 2017), since damage to serotonergic pathways in mice has only been detected following high doses of MDMA (Górska et al., 2018).For example, MDMA has been found to reduce striatal levels of dopamine, DAT, and TH, and to produce a persistent loss of dopaminergic cell bodies in the SNc of mice (Granado et al., 2008; Costa et al., 2017; Miner et al., 2017).Of note and consistent with prior data, abnormal activation of D1R and D2R seems to play a pivotal role in mediating MDMA-induced neurotoxic effects on the dopaminergic system of mice (Granado et al., 2011,2014).A partial sprouting of TH- and DAT-positive fibers has been observed in the CPu of MDMA-treated mice starting from the third day following drug administration, which may suggest that dopaminergic damage induced by MDMA in this brain area may recover over time (Granado et al., 2008).Interestingly, age and sex are two factors known to affect MDMA neurotoxicity in experimental animals.In this respect, two recent investigations provide new evidence, which may account for such differential susceptibility to the noxious effects of MDMA (Chitre et al., 2020; Costa et al., 2021).Adolescent mice have a higher metabolic turnover of dopamine following MDMA compared to adult mice (Chitre et al., 2020; Additional Table 4), and MDMA-treated male mice display an increased expression of the superoxide dismutase (SOD) type 2 enzyme in striatal dopaminergic terminals as well as a reduced striatal proteolytic activity when compared to female mice (Costa et al., 2021; Additional Table 4).
Other studies indicate that the neurotoxic effects of MDMA may extend beyond the dopaminergic system.Indeed, a decrease in the number of GABAergic interneurons has been detected in the hippocampus of rats subjected to repeated MDMA exposure (Anneken et al., 2013).In line with this finding, chronic intermittent MDMA treatment has been reported to lower the density of glutamic acid decarboxylase 67-positive fibers, an index of neurotoxicity affecting the GABAergic system, in the hippocampus, CPu,and mPFC of mice (Costa et al., 2017; Additional Table 4).
Recent studies have also shown that MDMA treatment affects the NO system.Indeed, acute administration of MDMA in rats was found to increase the expression of the inducible nitric oxide synthase, but not that of the endothelial or neuronal nitric oxide synthase, in the PFC (Schiavone et al.,2019; Additional Table 4).In the same cortical area, the authors also reported a significant elevation in immunoreactivity for 3-nitrotyrosine specifically in DAT-positive neurons, but not in glial cells (Schiavone et al., 2019).These observations are relevant, as increased 3-NT levels have been also documented in major neurodegenerative diseases, such as PD, Alzheimer’s,and Huntington’s diseases, and are thought to reflect the presence of massive nitrosative stress (Bandookwala and Sengupta, 2020).Moreover,in vitroandin vivoinvestigations have demonstrated that pharmacological inhibition of NO synthesis significantly decreases serotonergic and dopaminergic neurotoxicity induced by MDMA (García-Pardo et al., 2022).Nevertheless,clinical validation of these findings is still needed.
In humans, chronic MDMA use has been associated with the appearance of enduring neurochemical and functional alterations in serotonergic pathways.Reduced levels of serotonin and its major metabolite, 5-hydroxyindoleacetic acid, have been detected in the CPu (Kish et al., 2000) and cerebrospinal fluid(McCann et al., 1999) of MDMA users.PET investigations in MDMA users have demonstrated a significant reduction in the availability of SERT in multiple cortical brain regions and in the hippocampus (Kish et al., 2010; Roberts et al.,2016), as well as an increase in 5-HT2A receptor density in the occipital cortex(Reneman et al., 2000).Interestingly, a recent meta-analysis of 10 PET/SPECT studies in MDMA users reported that the reduction in SERT density was not directly connected with lifetime episodes of MDMA use (Müller et al., 2019).This suggests that other factors, such as the amount of drug taken on each occasion, should be taken into account during the evaluation of potential drivers of MDMA-induced neurotoxicity.This consideration is especially relevant with respect to MDMA’s evolving role as a therapeutic agent for the treatment of post-traumatic stress disorder.Furthermore, consistent with observations in nonhuman primates (Yeh et al., 2022), Müller et al.(2019)also reported a partial region-specific recovery in markers of serotonergic function after drug abstinence.Once more, this suggests that the noxious central effects of MDMA may be reversible to some extent (Müller et al.,2019).Nevertheless, additional studies are required to ascertain whether the restoration of SERT density indicates a full functional recovery of the serotonergic system.
A few studies have also investigated the presence of white matter alterations in MDMA users (De Win et al., 2008; Liu et al., 2011), with conflicting results(Roberts et al., 2018).A recent study found an elevation of FA in the corpus callosum and corticospinal tract of chronic MDMA users, which negatively correlated with MDMA use frequency (Zimmermann et al., 2022; Additional Table 4).While these results exclude the presence of a severe white matter lesion, they nevertheless suggest that MDMA use may cause abnormalities in axonal organization (Zimmermann et al., 2022).
Outside of changes in the central nervous system, a single oral dose of MDMA to human subjects increased plasma levels of cortisol and of inflammatory mediators (e.g.hydroxyeicosatetraenoic acid), and reduced levels of calcitriol(vitamin D), a hormone involved in the production of trophic factors (Boxler et al., 2018; Additional Table 4).In summary, data collected from human users suggest that MDMA induces changes in brain function primarily impacting the serotonergic system.Nonetheless, it remains unclear whether such changes reflect the presence of neurodegeneration in serotonergic pathways or neuroadaptive responses (Biezonski and Meyer, 2011).Moreover, it is important to state that most of the findings described here have been obtained from polydrug users, while studies involving pure/primary MDMA users are quite limited in number (Wunderli et al., 2017).Therefore, our knowledge of the precise effects, associated risks, and potential benefits of MDMA use remains to be further elucidated.
Methylphenidate
When taken orally according to a defined therapeutic plan, methylphenidate has negligible abuse potential and may elicit psychostimulant-like and sympathomimetic-like side effects of moderate intensity (e.g., repetitive movements, loss of appetite, insomnia, increase in blood pressure and heart rate) that can be controlled by dose adjustment.Nevertheless, concerns have been raised about possible long-term consequences that may arise from methylphenidate exposure during childhood and adolescence, which are two crucial phases of brain development (Loureiro-Vieira et al., 2017).In this regard, it has been estimated that globally, the 8–17% of college students aged 18–24 have consumed methylphenidate at least once without a valid medical indication, with rates of misuse reaching up to 35% depending on the country considered (Sharif et al., 2021; Zahavi et al., 2023).The primary incentives driving the misuse of methylphenidate include cognitive and academic enhancement, heightened alertness, weight loss, and personal curiosity (Finger et al., 2013).Since the chemical structure and mechanism of action of methylphenidate are similar to AMPH, prolonged (chronic) use of methylphenidate raises concerns about its neurotoxicity.Early studies in cell cultures and experimental animals provide conflicting results, since the neurotoxic effects of methylphenidate have been demonstrated by some (Gopal et al., 2007; Schmitz et al., 2016), but not others (Yuan et al.,1997; Ludolph et al., 2006).In addition, there is evidence to indicate that methylphenidate, administered after METH treatment in rats, prevents the persistent dopamine deficits and reverses the acute decreases in vesicular dopamine uptake and content and VMAT-2 ligand binding observed following METH alone (Sandoval et al., 2003).However, more recent investigations in rats have demonstrated that chronic methylphenidate may elicit neurotoxic effects (Motaghinejad et al., 2017a, b; Motaghinejad and Motevalian,2022) and dysfunctions in the ventral CPu (Quansah and Zetterström,2019; Additional Table 4).For example, chronic administration of methylphenidate reduced the number of granular cells in the dentate gyrus and pyramidal cells in the cornu ammonis region 1 (CA1) of the hippocampus(Motaghinejad et al., 2017b; Additional Table 4).These cellular reductions were accompanied by decreased expression of brain-derived neurotrophic factor, cAMP response element-binding protein (total and phosphorylated),protein kinase B (total and phosphorylated) and B-cell lymphoma-2 (Bcl-2)proteins, as well as increased expression of pro-apoptotic marker Bcl-2-like protein 4 (Bax) and glycogen synthase kinase 3 proteins in the hippocampus(Motaghinejad et al., 2017a, b; Additional Table 4).Furthermore, the same group found that methylphenidate altered markers of oxidative stress, such as malondialdehyde, glutathione, glutathione disulfide, and SOD, as well as markers of inflammation, such as interleukin-1β and tumor necrosis factor-α, in the hippocampus and/or mitochondria (Motaghinejad and Motevalian, 2022; Additional Table 4).Overall, these observations suggest the existence of pro-apoptotic, pro-oxidant, and pro-inflammatory effects of chronic methylphenidate (Motaghinejad et al., 2017a, b; Motaghinejad and Motevalian, 2022), and thereby raise concerns about its potential neurotoxic effects in humans who use methylphenidate chronically.However, it is important to note that these studies, which demonstrated neurotoxicity by chronic methylphenidate in rats, used a dose (10 mg/kg) that far exceeds therapeutic doses of methylphenidate, which are roughly 10 mg/day in children and 20-30 mg/day in adults, as indicated by the U.S.Food and drug administration guidelines.These doses correspond to 0.3–0.4 mg/kg,if an average weight of 32 kg or 70 kg is assumed for children and adults,respectively.Indeed, the available evidence from studies of structural and functional neuroimaging in unmedicated and medicated patients with ADHD indicates that methylphenidate is safe overall and improves brain function(Loureiro-Vieira et al., 2017; Krinzinger et al., 2019).Nevertheless, the same studies also indicate that brain toxicity (e.g., cerebral arteritis, hallucinations)may be induced by methylphenidate, although this effect seems to disappear after drug discontinuation (Loureiro-Vieira et al., 2017; Krinzinger et al., 2019).Overall, there is a substantial lack of data on the long-term neurological evaluation of patients treated therapeutically with methylphenidate(Krinzinger et al., 2019).Similarly, only a few studies have explored the detrimental effects of low doses of methylphenidate in experimental animals.Hence, further studies on the neurotoxic effects of methylphenidate in humans are warranted.Special focus should be placed on individuals who use methylphenidate for recreational purposes or as a performance enhancer,since they may be exposed to amounts of methylphenidate that far exceed therapeutic doses, which could result in overt neurotoxic effects.
Non-Sympathomimetic Psychostimulants
Non-sympathomimetic psychostimulants possess dissimilar chemical structures and include caffeine and nicotine.Caffeine is a methylxanthine alkaloid, which at doses used during recreational consumption, induces its central effects by antagonizing adenosine A1 and A2A receptors (Ferré et al., 2018).Caffeine also indirectly modulates other neurotransmitter systems, including dopaminergic pathways.This action contributes to the full manifestation of caffeine’s central effects as well as to its interactions with other psychoactive substances (Simola et al., 2021).Nicotine is a dinitrogen alkaloid that activates presynaptic nicotinic acetylcholine receptors, thus stimulating acetylcholine release (Benowitz, 2009).Nicotine also activates the dopaminergic system by increasing dopamine release in mesolimbic regions,CPu, and PFC (Benowitz, 2009).In addition to eliciting psychostimulant effects, caffeine, and nicotine may both induce neurochemical alterations possibly leading to brain dysfunctions and/or neurotoxicity.
Caffeine
Caffeine is a natural xanthinic alkaloid contained in several plant species(e.g.,Coffea arabica,Thea sinensis) and consumed worldwide due to its mild euphoriant and psychostimulant effects.According to the latest European Food Safety Authority (EFSA) dietary survey, the daily intake of caffeine in Europe (expressed as mg/kg × body weight) ranges from 0.1 to 1.4 in adolescents (10–17 years old), from 0.5 to 4.3 in adults (18–65 years old), and from 0.3 to 4.8 in the elderly (60–74 years old) (Verster and Koenig, 2018).For adults, major sources of caffeine are coffee and tea, followed by carbonated soft drinks, while adolescents and children mostly consume carbonated softdrinks and tea, followed by coffee [EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA), 2015].
Caffeine consumption is generally considered a harmless habit, which, in some cases, may even be beneficial for an individual’s performance or health.For example, caffeine has been shown to be protective in experimental models of neurotoxicity (Ren and Chen, 2020).In addition, epidemiological studies suggest that caffeine-mediated neuroprotection could occur in humans, since there is an inverse correlation between caffeine consumption and the incidence of neurodegenerative diseases (Janitschke et al., 2021).Nevertheless, harmful effects associated with caffeine consumption have been also described.For example, caffeine may amplify the behavioral effects of dopaminergic psychostimulants of abuse, such as AMPH-related drugs and cocaine, in both experimental animals and humans (Morelli and Simola,2011).Furthermore, preclinical studies have demonstrated that caffeine may elicit neurotoxic effects and exacerbate the neurotoxicity elicited by other substances (Frau et al., 2016).
Earlier investigations that employedin vitroorin vivomodels demonstrated that caffeine-induced neurotoxicity may be sustained by diverse mechanisms,such as induction of apoptosis (Kang et al., 2002), alteration in the cellular levels of Ca2+(Gepdı̇Remen et al., 1998), inhibition of neuronal repair and induction of neuroinflammation (Yang and Jou, 2016), inhibition of SOD and catalase enzymes and induction of lipid peroxidation (Bavari et al., 2016;Figure 1).On the other hand, activation of necrotic and excitotoxic pathways is not involved in caffeine-mediated neurotoxicity (Kang et al., 2002),whereas the involvement of oxidative stress has been suggested by some studies (Bavari et al., 2016), but not others (Kang et al., 2002).More recentin vitroinvestigations have shed light on the mechanisms by which caffeine induces neurotoxicity and how it could amplify the neurotoxic effects of other substances.A study in PC12 cells provides support to the involvement of oxidative stress pathways in caffeine-mediated neurotoxicity, by demonstrating that concentrations of caffeine that inhibited cell viability also induced apoptosis and increased the generation of ROS (Chian et al., 2022;Additional Table 5).Moreover, caffeine treatment reduced protein expression and mRNA levels of Nrf2 protein, a key regulator of oxidative stress response,as well as of the mRNA encoding Nrf2 and its target genes NADPH quinone oxidoreductase 1, glutamate cysteine ligase catalytic subunit and modifier subunit (Chian et al., 2022; Additional Table 5).Further support to the role of the Nrf2 pathway in the toxic effects of caffeine comes from the finding that caffeine treatment had a reduced effect on the viability of PC12 cells bearing a knockdown of Nrf2 expression (Chian et al., 2022; Additional Table 5).A study performed in SH-SY5Y cells demonstrated that concentrations of caffeine within the range of those detected in caffeine-contaminated environments (e.g., wastewater, seawater) under-expressed several genes encoding key elements of other neurotransmitter pathways, such as the serotonin 5HT3A receptor, the D2R, the enzyme GABA-transaminase, the protein synaptotagmin 10, which regulates neurotransmitter release, and the subunit a3 of the ATPase Na+/K+pump (Vulin et al., 2022; Additional Table 5).Although this study found no overt cytotoxicity in SH-SY5Y cells exposed to the concentrations of caffeine tested (Vulin et al., 2022), the observed changes in gene expression deserve further consideration, since they identify new molecular targets that could potentially sustain caffeine’s neurotoxic effects through prolonged exposure to low amounts or acute exposure to high amounts of caffeine.
Additional studies have identified the induction of apoptotic cell death as a key mechanism by which caffeine potentiates the neurotoxic effects of other substances.A study in SH-SY5Y cells exposed to ethanol found that subsequent treatment with caffeine exacerbated the reduction in cell viability and increased early apoptosis (Sangaunchom and Dharmasaroja, 2020).Specifically, caffeine potentiated ethanol-induced inhibition of signaling mediated by mechanistic target of rapamycin, p70S6 kinase, and eukaryotic translation initiation factor 4E-binding protein 1, and eventually reduced mitochondrial membrane potential, as compared to either control conditions or ethanol alone (Sangaunchom and Dharmasaroja, 2020; Additional Table 5).Moreover, an investigation in premature newborn mice found that the co-administration of caffeine together with the sedative/anesthetic drugs midazolam, fentanyl, or ketamine potentiated the number of activated caspase 3 apoptotic cells in colliculi, hippocampus, neocortex, CPu, and thalamus, as compared to treatment with either drug alone (Cabrera et al., 2017; Additional Table 5).Caffeine has also been shown to exacerbate apoptosis induced by morphine in the premature rat brain (Kasala et al.,2020).In this model, combined administration of caffeine and morphine,compared to either drug alone, caused a more marked and sex-dependent increase in the expression of Bax and a more marked reduction in the expression of Bcl-2 (Kasala et al., 2020; Additional Table 5).Furthermore,caffeine has been reported to worsen the neurotoxicity induced byin uteroexposure to isoflurane anesthesia in the fetal rhesus macaque brain(gestational days 100–120), by exacerbating neuronal apoptosis, especially in the basal ganglia and cerebellum (Noguchi et al., 2018; Additional Table 5).Interestingly, the brains of rhesus macaques prenatally exposed to isoflurane and caffeine displayed a decreased number of oligodendrocytes throughout their white matter (Noguchi et al., 2018; Additional Table 5).
It appears that mechanisms other than induction of apoptotic cell death may participate in the amplification of drug-induced neurotoxicity by caffeine.For example, caffeine potentiates MDMA-induced damage in dopaminergic and serotonergic terminals in the mouse brain following binge co-administration of caffeine and MDMA (Górska et al., 2018).This effect was associated with an increase in extracellular dopamine and serotonin levels in the CPu and an exacerbation of the oxidative damage of nuclear DNA induced by MDMA in the mPFC, compared to either drug alone (Górska et al., 2018; Additional Table 5).Another study found that co-administration of caffeine and cocaine,compared to either drug alone, exacerbated the increase in intracellular Ca2+and dysregulated the hyperpolarization-activated cyclic nucleotide-gated and T-type VGC channels in thalamic ventrobasal neurons of mice (Rivero-Echeto et al., 2021; Additional Table 5).Although this study did not assess the neurotoxic effects of the caffeine-cocaine combination, the observed increase in intracellular Ca2+suggests the existence of other mechanisms by which caffeine could potentiate the neurotoxic effects of other drugs, since dysregulation of Ca2+homeostasis is a key player in neurotoxicity phenomena.
Despite evidence that caffeine may induce neurotoxicity and can exacerbate the neurotoxic effects of other substances, the neuroprotective effects of caffeine have also been extensively demonstrated (Simola et al.,2021).Various factors may influence the induction of neuroprotection/neurotoxicity by caffeine, including levels of caffeine exposure, the presence of interindividual and interspecies differences in caffeine pharmacokinetics and metabolism, as well as other drugs with which caffeine is consumed.Caffeine acts as an antagonist of adenosine A1 and A2A receptors at plasma concentrations attained following recreational consumption (Fredholm et al., 1999).In this regard, it is noteworthy that several of the studies that demonstrated caffeine neurotoxicity used concentrations/doses that exceeded those associated with the recreational consumption of caffeine.As such, these high concentrations/doses of caffeine may act on targets other than adenosine receptors (Fredholm et al., 1999), and, in turn, may activate molecular pathways that cause neurotoxicity.Nevertheless, the potential of caffeine to induce, or exacerbate, neurotoxicity should be regarded as a source of concern at least under specific circumstances, such as the use of high doses of caffeine for the treatment of infants for apnea of prematurity(Noguchi et al., 2018) and its recreational consumption in combination with other psychoactive substances that bear neurotoxic potential (Frau et al.,2016).Furthermore, the increasing presence of caffeine and its metabolites as environmental contaminants raises concern regarding potential neurotoxicity following the consumption of caffeine-contaminated food and water (Vieira et al., 2022).
Nicotine
Nicotine is a natural alkaloid contained in plant species from the genusNicotianaand is consumed worldwide due to its euphoriant properties.It is found in several products, such as cigarettes, e-cigarettes, and nicotine cessation gums and patches.In 2019, more than 1 billion people worldwide were reported to regularly smoke cigarettes, and almost 8 million deaths were attributable to cigarette use (GBD 2019 Tobacco Collaborators).Nicotine elicits its euphoriant and psychostimulant properties by activating nicotinic acetylcholine receptors in the brain, which causes the release of a variety of neurotransmitters including norepinephrine (Verplaetse et al., 2015),acetylcholine (Wonnacott, 1997), serotonin (Fletcher et al., 2008), GABA and glutamate (Li et al., 2014), and dopamine (Di Chiara, 2000).The release of these neurotransmitters, in particular of dopamine, serves as the basis of the psychoactive, rewarding, and addictive properties of nicotine (Di Chiara,2000).Several pathological mechanisms have been implicated in the brain dysfunctions that can be observed following nicotine use, including, but not limited to, apoptosis (Anbarasi et al., 2006), bioenergetic failure (Malińska et al., 2019), and neuroinflammation (Piao et al., 2009; Figure 1).
Multiple factors influence the long-term effects of nicotine, leading to either brain dysfunctions or neuroprotection depending on the age at which it is consumed (Ren et al., 2022), the amount consumed, and the duration of its use (Peng et al., 2018).For example, nicotine use during adulthood is associated with increased risk for psychiatric and neurological conditions,including major depression (Laviolette, 2021), alcohol use disorder (King and Meyer, 2022), lower processing speed, poorer general cognitive ability,poorer decision-making, increased impulsivity (Contiand Baldacchino, 2021,2022), and Alzheimer’s disease (Durazzo et al., 2018).It may be hypothesized that the increased risk of brain disease in nicotine users may depend, at least in part, on the induction of abnormalities in cerebral structure and function,as suggested by neuroimaging studies (see below).Conversely, preclinical and clinical evidence indicates that nicotine administration/consumption may have a protective effect on the underlying neurodegeneration and/or phenotypical manifestation of PD (Nicholatos et al., 2018; Mappin-Kasirer et al., 2020; Carvajal-Oliveros et al., 2021; Wang et al., 2022).
Nicotine is often consumed by pregnant women, and gestational nicotine exposure has been shown to negatively affect the developing fetal brain,leading to neuronal and glial alterations in several cerebral regions that may affect the offspring later in their life (Dwyer et al., 2019; Zhou et al., 2021).For example, a recent study demonstrated the existence of abnormalities in cortical regions of adolescent rats that were exposed to nicotinein uterofrom gestational days 4 to 18 (Dwyer et al., 2019; Additional Table 5).Specifically,male and female adolescent rats prenatally exposed to nicotine displayed an increase in dopamine content, a decrease in the turnover ratio of homovanillic acid/dopamine, and a decrease in norepinephrine transporter expression in the PFC, compared to adolescent rats not prenatally exposed to nicotine(Dwyer et al., 2019; Additional Table 5).There was also an increase in striatal and pallidal D3R binding, as measured by [125I]-7-OH-PIPAT, and D2R-Gprotein functional coupling in the ventral CPu and pallidum in adolescent rats prenatally exposed to nicotine (Dwyer et al., 2019; Additional Table 5).In the same study, adolescent female rats prenatally exposed to nicotine exhibited a decrease in striatal DAT binding, as measured by [125I] RTI-55 (Dwyer et al.,2019; Additional Table 5).A different study using mice that were prenatally exposed to nicotine reported an increased number of microglial cells and Nissl-positive neurons in the CA1 of the hippocampus, as well as alterations in the expression of genes related to neuroinflammation, synaptic plasticity,and neurotransmitter function, compared to mice prenatally treated with saline (Zhou et al., 2021; Additional Table 5).Nicotine exposurein uterocan also affect an offspring’s response to subsequent challenges with a different psychostimulant.For example, in male adolescent mice born to mothers treated with nicotine during pregnancy methylphenidate treatment led to more pronounced release of dopamine and noradrenaline, but not serotonin,in the PFC (measured byin vivomicrodialysis), compared to mice prenatally exposed to saline (Zhang et al., 2021; Additional Table 5).
Neuroimaging investigations in humans have obtained evidence supporting the possibility thatin uteroexposure to nicotine may elicit enduring detrimental effects on brain function in offspring.A recent MRI study demonstrated the presence of smaller volumes of the left and right thalami and inferior frontal gyrus in children aged 7–9 years old who were prenatally exposed to environmental tobacco smoke, compared to age-matched children not exposed to tobacco smoke during gestation (Margolis et al., 2021;Additional Table 5).Another MRI study found lower callosal volume, higher FA, and lower mean diffusivity in adolescent and young adults prenatally exposed to maternal cigarette smoking, compared to age-matched nonexposed individuals (Björnholm et al., 2020; Additional Table 5).
Studies of neuroimaging performed in cigarette smokers have also demonstrated that chronic nicotine consumption during adulthood is associated with brain abnormalities in gray or white matter (Elbejjani et al., 2019; Yang et al., 2020; Fan et al., 2023).Specifically, Elbejjani et al.(2019) reported a reduction in gray matter volume in adult cigarette smoker participants, compared to adult non-smokers (Additional Table 5).In line with this, a recent meta-analysis of 17 voxel-based morphometry studies found that chronic smokers had a robust decrease in gray matter volume in the right superior frontal gyrus, right middle frontal gyrus, left insular and left superior frontal gyrus (Yang et al., 2020; Additional Table 5).Furthermore, reductions in the thickness of the medial and lateral orbitofrontal cortex, entorhinal cortex, fusiform and middle temporal gyrus, and insula have been observed in cigarette smokers, compared to people who never smoked (Durazzo et al.,2018; Additional Table 5).Conversely, Yang et al.(2020) found an increased gray matter volume in the right lingual cortex and left occipital cortex of cigarette smokers (Additional Table 5).In line with this, a more recent MRI study found that cigarette smokers had higher gray matter volume in the leftbut not right putamen, compared to non-smokers, whereas no differences in gray matter volume were observed in the putamen, compared to cannabis and tobacco smokers (Daniju et al., 2022; Additional Table 5).
Conclusions
Of late, much progress has been achieved in clarifying the central noxious effects of psychostimulants.Indeed, while consistent evidence supports the therapeutic applications of some psychostimulants (e.g., amphetamine,methylphenidate) under closely monitored conditions, it is now clear that their misuse may potentially result in brain impairments and functional neurotoxicity.
We have reviewed the recent findings derived from experimental models and human studies that examined brain dysfunctions and neurotoxicity that may be triggered by the most commonly used sympathomimetic and non-sympathomimetic psychostimulants.The preclinical investigations reported in this review provide new insights into factors that may contribute to psychostimulant-induced neurotoxicity.These data also substantiate previous findings, indicating that consumption of psychostimulants,especially at high amounts and for extended periods of time, could result in the emergence of neurotoxic phenomena and brain dysfunctions which involve monoaminergic systems.Of note, oxidative stress, mitochondrial dysfunction, neuroinflammation, and apoptosis dysregulation emerge as prominent and common mechanisms involved in the neurotoxic effects of psychostimulants.At the same time, each psychostimulant may have its own specific mechanism(s) that mediates its detrimental effects on the brain, as well as unique noxious interactions with other substances that may lead to/contribute to brain damage.
It is important to note that the extent of psychostimulant-induced neurotoxic effects varies and depends on the specific psychostimulant considered, the experimental procedure, as well as the stage of life at which it is consumed or administered.Furthermore, recent findings indicate that detrimental alterations outside the central nervous system, for example in the gut microbiota, are of particular interest since they may provide new tools for monitoring the noxious effects of psychostimulants in users.Conversely,data derived from neuroimaging investigations in users provide compelling evidence that psychostimulants may elicit central structural and functional changes that impact the activity of several cortical and subcortical brain areas;interestingly, some of these changes appear to undergo recovery following extended periods of drug abstinence.Nevertheless, the question remains as to whether the psychostimulant-induced alterations observed in neuroimaging studies reflect the occurrence of neuronal cell death or should be attributed to adaptive responses triggered by aberrant monoaminergic signaling.Moreover, potential confounding variables such as polydrug use, inclusion of current or abstinent users, drug intake patterns, and amount of drug taken per occasion, introduce complexity into the interpretation of clinical studies.In this regard, further research involving animal models designed to more accurately replicate the patterns of drug consumption observed in humans will be crucial for gaining novel insights into the neurotoxic effects and brain dysfunction that psychostimulants may induce in the human brain.
Author contributions:MS wrote the parts on toxicity induced by amphetamine and its derivatives.NS wrote the parts on toxicity induced by methylphenidate and caffeine.GC wrote the parts on toxicity induced by cocaine and nicotine.MS and GC created the tables and figure.All authors critically revised the whole manuscript and approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Data availability statement:All data relevant to the work are included in the article or uploaded as Additional files.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional files:
Additional Table 1: Classification of psychostimulants.
Additional Table 2: Recent preclinical and clinical studies evaluating amphetamine (AMPH)-and cocaine-induced brain dysfunctions and neurotoxicity.
Additional Table 3: Recent preclinical and clinical studies evaluating methamphetamine (METH)-induced brain dysfunctions and neurotoxicity.
Additional Table 4: Recent preclinical and clinical studies evaluating 3,4-methylenedioxymethamphetamine (MDMA)-and methylphenidateinduced brain dysfunctions and neurotoxicity.
Additional Table 5: Recent preclinical and clinical studies evaluating caffeineand nicotine-induced brain dysfunctions and neurotoxicity.

- 中国神经再生研究(英文版)的其它文章
- NADPH oxidase 4 (NOX4) as a biomarker and therapeutic target in neurodegenerative diseases
- Circadian rhythm disruption and retinal dysfunction:a bidirectional link in Alzheimer’s disease?
- Interplay between the glymphatic system and neurotoxic proteins in Parkinson’s disease and related disorders: current knowledge and future directions
- Roles of neuronal lysosomes in the etiology of Parkinson’s disease
- Therapeutic advances in neural regeneration for Huntington’s disease
- The advantages of multi-level omics research on stem cell-based therapies for ischemic stroke