
Highly toxic Aβ begets more Aβ
2024-01-10 10:07MercKemehNoelLazo
中国神经再生研究(英文版) 2024年9期
Merc M.Kemeh, Noel D.Lazo
Alzheimer’s disease (AD) is the most common form of dementia-the 7thleading cause of death worldwide.At the tissue level, AD is characterized by the presence of extracellular amyloid plaques that are comprised primarily of the amyloid-β peptide (Aβ), and by intraneuronal neurofibrillary tangles composed of tau.Molecular genetics of early-onset AD and longitudinal brain-imaging studies of late-onset AD indicate that extracellular Aβ deposition, in general, precedes neurofibrillary tangle formation in neurons (Hampel et al., 2021;Young-Pearse et al., 2023).
Aβ is produced by proteolytic processing of the Aβ precursor protein (AβPP) via the amyloidogenic pathway, that is, AβPP is cleaved consecutively by β- and γ-secretase to generate Aβ peptides of varying lengths, of which the two predominant forms are Aβ1–40and Aβ1–42.Early-onset AD missense mutations in AβPP and presenilin – the catalytic subunit of γ-secretase – affect AβPP processing by increasing the production of Aβ1–42relative to Aβ1–40.Recently, a polygenic risk score for late-onset AD has also been associated with increased production of Aβ1–42(Lagomarsino et al., 2021).Aβ1–42is more neurotoxic than shorter Aβ variants, as revealed by many studies usingin vitroandin vivosystems.Moreover, Aβ1–42, under conditions that are not well understood, undergoes sequential post-translational modifications by dipeptidyl peptidase 4 and glutaminyl cyclase(QC) to produce AβpyroE3–42(Figure 1).Compared to Aβ1–42, AβpyroE3–42is more hydrophobic, which in turn, makes it more insoluble and amyloidogenic(Bayer, 2022).AβpyroE3–42is a major component of diffuse and compacted amyloid plaques in AD.Importantly, AβpyroE3–42has been shown to cause template-induced misfolding of Aβ1–42into oligomers that spread in a prion-like fashion.In vitroandin vivostudies have shown that the AβpyroE3–42-seeded oligomers are highly neurotoxic,synaptotoxic, and proinflammatory, suggesting that AβpyroE3–42even when present in small amounts contributes significantly to early neuronal cell death in AD.
Nature has provided quality control mechanisms for clearing Aβ from the brain.These include enzymatic degradation, phagocytosis by glial cells, transport across the blood-brain barrier,and clearance mediated by the bulk flow of cerebrospinal and interstitial fluids (Tarasoff-Conway et al., 2015).The relative importance of these mechanisms has not yet been established.Nonetheless, increasing evidence suggests that enzymatic degradation of Aβ monomer is an important and efficient mechanism for Aβ clearance.
The insulin-degrading enzyme (IDE; EC 3.4.24.56)is a conserved and ubiquitous zinc metalloprotease that could be the most important enzyme for degrading Aβ monomer in the brain (Kurochkin et al., 2018).IDE hypofunction and hyperfunction increase and decrease brain levels of Aβin vivo,respectively.As its name suggests, IDE degrades α-helical insulin; however, IDE’s natural substrates are predominantly unstructured molecules that have a high propensity to form β-sheet, including Aβ and the islet amyloid polypeptide.IDE’s catalytic activity is allosterically regulated by small molecules such as ATP (Im et al., 2007), carnosine(Distefano et al., 2022), and short peptides (Song et al., 2003).
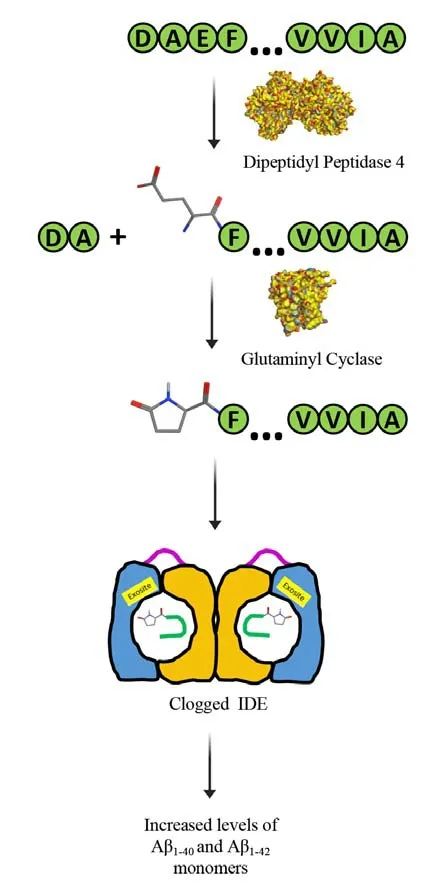
Figure 1 |Highly toxic AβpyroE3–42 begets more Aβ.Aβ1–42 is modified to AβpyroE3–42 by the sequential action of dipeptidyl peptidase 4 (PDB ID: 1NU6) and glutaminyl cyclase (PDB ID: 2AFM).AβpyroE3–42, arguably the most toxic form of Aβ, deactivates the insulin-degrading enzyme by clogging it up.In turn, the loss of IDE activity leads to increased brain levels of Aβ1–40 and Aβ1–42.Created using Microsoft PowerPoint.Aβ: Amyloid-β; IDE: insulindegrading enzyme.
Recently, we found that AβpyroE3–42inhibits IDE’s activity towards Aβ1–40(Kemeh and Lazo, 2023).A plausible explanation for this inhibition is the co-aggregation of Aβ1–40with AβpyroE3–42, secluding the former from IDE.Given that AβpyroE3–42also inhibits the IDE-dependent degradation of insulin(Kemeh and Lazo, 2023), a more reasonable mechanism is that the modified Aβ peptide causes IDE to become inactive.X-ray-crystallographic and cryogenic-electron microscopic studies have revealed key structural and enzymological features of the enzyme (Tang, 2016; Zhang et al., 2018).IDE’s functional form is a homodimer, with each monomer composed of two bowl-shaped halves held together by a flexible linker.Furthermore,each monomer can exist in two dominant conformations: an open conformation for the capture of substrate and release of products; and,a closed conformation to form a catalytic chamber(aka crypt) for proteolysis to occur.Because the substrate must fit within IDE’s crypt, substrates are limited to peptides that contain less than 80 residues.IDE contains a highly conserved exosite that presumably interacts with the N-terminus of its substrate through electrostatic interactions(Tang, 2016; Ivancic et al., 2018), facilitating the substrate’s unfolding prior to its degradation.We posit that because AβpyroE3–42’s N-terminus is uncharged, its exosite-assisted unfolding does not occur.As a result, AβpyroE3–42clogs up each“clamshell” of IDE, and by doing so, the enzyme becomes deactivated (Kemeh and Lazo, 2023;Figure 1).Our finding implicates AβpyroE3–42in the reduction of IDE activity which has been associated with late-onset AD.
Could the clogging up of IDE by AβpyroE3–42be prevented by small molecules? Maybe.Small molecules such as ATP (Patel et al., 2017) and polyphenols (Zheng et al., 2019) could modulate the interactions between AβpyroE3–42and IDE in such a way that the Aβ peptide does not clog up IDE’s crypt.ATP has properties of biological hydrotrope in that it has the ability to inhibit the formation of protein aggregates and keep proteins in monomeric form.Polyphenols possess the ability to modulate protein-protein interactions found in amyloidogenic assemblies.Interestingly,resveratrol a polyphenol found in red wine also sustains IDE activity towards Aβ1–42(Krasinski et al., 2018).Could the formation of AβpyroE3–42be inhibitedin vivoby small molecules? Of the two enzymes implicated in the conversion of Aβ1–42to AβpyroE3–42(Figure 1), QC appears to be the more attractive target for inhibitor development.We speculate that as long as the N-terminus of Aβ is negatively charged, as would be in the case in Aβ3–42that is produced by N-terminal truncation of Aβ1–42by dipeptidyl peptidase 4, the peptide would be susceptible to degradation by IDE.Furthermore,pyroglutamate formation in truncated proteins such as α-synuclein and TDP-43 could contribute to co-occurring pathologies in AD, providing an additional rationale for inhibiting QC.Of the QC inhibitors under development, varoglutamstat has advanced to Phase 2a/b trials in the US.The hope is that this small molecule could be an alternative to monoclonal antibodies targeting AβpyroE3–42or Aβ1–42, which have been shown to cause amyloidrelated imaging abnormalities in some patients.
Finally, our finding that a highly toxic Aβ begets more Aβ by deactivating IDE also brings to the forefront the development of small molecules that could increase IDE’s activity towards a particular substrate.Because IDE is an allosteric enzyme,and significant progress has been made in the elucidation of its structural biology, we believe that the stage is now set for the development of IDE activators that specifically target the enzyme’s inactivity towards the most toxic form of Aβ.
This work was supported by the National Institutes of Health through grant R15AG055043 to NDL and also by the Lise Ann and Leo Beaver’s endowment to Clark University.
Merc M.Kemeh, Noel D.Lazo*Gustaf H.Carlson School of Chemistry and Biochemistry, Clark University, Worcester, MA, USA
*Correspondence to:Noel D.Lazo, PhD,NLazo@clarku.edu.
https://orcid.org/0000-0003-1769-7572(Noel D.Lazo)
Date of submission: September 14, 2023
Date of decision: October 26, 2023
Date of acceptance: November 8, 2023 Date of web publication: December 15, 2023
https://doi.org/10.4103/1673-5374.390983
How to cite this article:Kemeh MM, Lazo ND(2024) Highly toxic Aβ begets more Aβ.Neural Regen Res 19(9):1871-1872.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- NADPH oxidase 4 (NOX4) as a biomarker and therapeutic target in neurodegenerative diseases
- Circadian rhythm disruption and retinal dysfunction:a bidirectional link in Alzheimer’s disease?
- Interplay between the glymphatic system and neurotoxic proteins in Parkinson’s disease and related disorders: current knowledge and future directions
- Roles of neuronal lysosomes in the etiology of Parkinson’s disease
- Therapeutic advances in neural regeneration for Huntington’s disease
- The advantages of multi-level omics research on stem cell-based therapies for ischemic stroke