
New complex physiological findings evolve hypothesized mechanisms of Dravet syndrome
2024-01-10 10:07MacKenzieHoward
中国神经再生研究(英文版) 2024年9期
MacKenzie A.Howard
Developmental and epileptic encephalopathies(DEEs) are neurological disorders generally involving medically intractable seizures and a diverse array of comorbid neuropsychiatric delays and deficits that may severely affect cognition,mood, sleep, social behavior, movement control,learning, and/or memory.Dravet syndrome(DS), also known as Severe Myoclonic Epilepsy of Infancy, is a rare disease but one of the more common DEEs, afflicting children in infancy and causing severe lifelong struggles and high risk of early mortality (Villas et al., 2017).Most people with DS have a deleterious variant to one copy of theSCN1Agene, which encodes the voltagegated sodium channel Nav1.1.Variants ofSCN1B,HCN1, and other genes have also been identified as potentially causative in diagnosed cases of DS.Animal models across phyla with changes to the expression ofScn1aor its ortholog present with seizures, neurological deficits, and early mortality analogous to DS.Early animal model work provided a wealth of knowledge about the role of Nav1.1 in neural excitability and dysfunction and brought about the hypothesis that GABAergic interneuron hypoexcitability was the underlying cause of DS.Here we provide a brief review of some recent studies that broaden the interneuron hypothesis, revealing cell phenotypes and circuit interactions that are subtle, complex, and at times counterintuitive, all while deepening our understanding of the intricate nature of neural processing and the mechanisms of neurological disease.
Given the foundational importance of voltagegated sodium channels in neuronal excitability,how loss of such a channel would lead to the hyperactivity associated with epilepsy only becomes clear when cell-type specificity is considered.The original report onScn1a+/–mice(i.e., haploinsufficent, which best mimics the genetic variants of many people with DS) revealed decreases in voltage gated sodium currents and action potential firing in hippocampal GABAergic interneurons compared toScn1a+/+controls,but no such change in excitatory pyramidal neurons (Yu et al., 2006).This birthed the interneuron hypothesis for DS, supposing that decreased excitability of these cells would result in decrements in synaptic inhibition and loss of inhibitory control over circuit activity, thus leading to seizures and other neuropsychiatric changes(Figure 1A).These initial findings were followed up with many detailed studies using constitutive and cell-type specific haploinsufficiency ofScn1athat were highly focused on parvalbumin- (PV)and somatostatin-positive interneurons in the hippocampus (well-reviewed previously by Catterall, 2018).Taken together, these studies show that Nav1.1 decrements indeed have cell type-specific effects, and removal ofScn1afrom specific cell types is sufficient to cause seizures and behavioral changes.Recently, several lines of evidence have revealed that cellular and circuit level changes in DS models are more complex than just reduced interneuron excitability (Figure 1B).For example, whileScn1a+/–PV+interneuron excitability is decreased during the first few postnatal weeks, including thetime of seizure onset in these mice, the excitability of this cell type then normalizes to wild type levels as the mice grow into adulthood, but seizures and behavioral deficits remain (Favero et al.,2018).While some interneuron subtypes continue to exhibit hypoexcitability into adulthood, this study demonstrates important points worthy of highlight.First, the onset of a disease state and the maintenance that a disease state can have different mechanisms.Second, the developing brain is not just a smaller version of the adult brain: neural differences in early development can have a different outcome on function than the same differences at later stages, and conversely,different sets of cellular phenotypes can produce similar changes in function at different stages of development.Third, unknown phenotypes,compensation, and/or plasticity may alter the neural circuit such that correcting the major known phenotype may not cure the disease state.Finally, rigorous testing of intuitive hypotheses often reveals more sophisticated mechanisms than originally expected.
In vivorecordings of interneurons and other celltype activity reinforced the complex connection betweenin vitrohypoexcitability and circuit function.UsingScn1ahaploinsufficient mice, De Stasi et al.(2016) reported a major finding that ran surprisingly counter to expectations built upon the first wave ofin vitrostudies and the interneuron hypothesis: the activity of cortical inhibitory neurons was not decreased in DS mice compared with wild type during normal ongoing activity.Using extracellular recordings, De Stasi showed that firing patterns and rates of individual PV and somatostatin-positive interneurons in the cortex were unchanged between DS and control mice in the interictal period (i.e., between seizures).They verified that these interneuron subtypes showed decreased intrinsic excitabilityin vitroin mice aged younger than P20.They then showed that optogenetically reducing the activity of these interneuron populationsin vivoinduced high-frequency activity in the cortical field potential, showing that inhibition was effectively controlling hyperactivity in the circuit.Intriguingly,theirin vitroexperiments also documented changes to synaptic excitation within the cortical circuitry, in addition to the expected decreases in synaptic inhibition.These data suggest that interneuron hypoexcitability in and of itself does not necessarily cause deficits in ongoing circuit processing and changes to excitatory connectivity,whether as primary or secondary phenotype,occur after decreases inScn1aexpression.
Further in-depth analysis of PV+interneuron activityin vivohas revealed that these neurons can be more activein vivodespite decreased intrinsic excitability.In a detailed study by Tran et al.(2020), cortical PV+interneurons and pyramidal neurons both showed higher baseline activity rates in awake behavingScn1a+/–mice compared to the control.In the DS model, firing rates of both populations of neurons were also higher than in the wild type, but synchrony of interneuron firing was decreased in the preictal phase before the onset of febrile-induced seizures.As in the De Stasi et al.(2016) study, these data show that decreased neural excitabilityin vitrodoes not map directly ontoin vivoactivity levels, and thus the mechanisms for neurological deficits lie in complex circuit interactions.
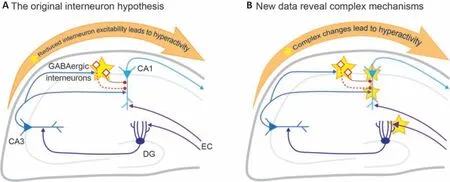
Figure 1 |New physiological findings in Scn1a-deficient mouse models of Dravet syndrome.Simplified hippocampal circuit illustrating the original interneuron hypothesis (A), in which the primary phenotype is reduced GABAergic interneuron excitability (altered physiology indicated by stars).Recent studies have revealed surprising new aberrant phenotypes within this circuity (B).Created with Adobe Illustrator.DG: Dentate gyrus; EC:inputs from Entorhinal cortex.
More recently, multiple groups have reported experiments attempting to tease apart these network interactions.Work from my lab sought to understand how interneuron hypoexcitability would change cellular information processing as pyramidal neurons integrate excitatory and inhibitory inputs.Examining CA1 pyramidal neuron input-output functions, we expected to find increased throughput in response to physiologically patterned theta burst stimulation(Chancey and Howard, 2022).Based on the interneuron hypothesis, we expected that loss of inhibitory tone would either cause greatly increased responses or would reveal evidence of homeostatic decreases in excitatory synaptic input strength that served to normalize outputs.Surprisingly, we found that neither of these expectations held true.Instead, subthreshold and suprathreshold responses to mixed excitatory and inhibitory synaptic input were unchanged inScn1a+/–pyramidal neurons compared with control.Neither were there decreases in excitatory synaptic currents indicating homeostatic downscaling due to loss of the gene or the seizures that these animals experience.We did,however, find changes to inhibitory synaptic release probability and increases in the temporal summation of inhibitory postsynaptic potentials.The mechanism causing these changes, or how they might be helping control circuit throughput,are not yet clear, but either of these changes could increase the efficacy of inhibition in the face of decreased interneuron excitability.Further work in CA1 pyramidal neurons by Jones et al.(2022) reported reduced intrinsic excitability in these neurons at early points in development near the onset of seizure activity.Interestingly,while decreases in pyramidal neuron excitability normalized in the third postnatal week, they do so just as the mice enter the age where mortality rates are highest in this model.Using a different stimulation technique than our own, they also report stable synaptic integration in CA1 across the developmental stages tested despite ongoing changes in both pyramidal and interneuron excitability.Both these reports highlight how the many interacting variables in even a simple circuit can produce outcomes counter to expectations,and how normalization of cellular phenotypes may not lead to normal brain outcomes.
Other recent studies highlight the importance of looking not just beyond interneurons, but beyond cortex and hippocampus.Importantly,the thalamus and thalamocortical processing are well known to be important in DS and many other epilepsy disorders.Ritter-Makinson et al.(2019) made the first detailed study of howScn1ahaploinsufficiency altered the physiology of thalamic neurons, using a transgenic haploinsufficiency mouse model expressing one copy of a deleterious humanSCN1Avariant.This study focused on thalamic reticular nucleus (nRT)neurons, which are GABAergic and PV+, but are projection neurons that can fire bursts or tonic trains of spikes, as opposed to the fast-spiking interneuron phenotype associated with most cortical and hippocampal PV+GABAergic cells.And unlike PV+interneurons,Scn1adeficiencies in nRT neurons caused increased excitability in the form of enhanced bursting, due to downregulation of calcium-activated potassium channels.This likely shifts interactions within the thalamocortical circuit and may thus drive hyperactivity and non-convulsive seizures.This novel mechanism provides further evidence that hidden changes secondary to the loss of Nav1.1 may play an active role in the DS disease process.
Further work in the thalamus by Studtmann et al.(2022) provided a detailed study of both inhibitory and excitatory neurons in the somatosensory corticothalamic loop.They reported fascinating contrasts in how the loss ofScn1achanges the physiology of different neurons in this circuit.Glutamatergic thalamocortical neurons of the ventral posteromedial nucleus showed increased tonic firing in the DS model, but no changes to burst firing.Conversely, ventral posterolateral glutamatergic thalamocorticalScn1a+/–neurons exhibited profound decreases in tonic firing but increases in burst firing.They also report that GABAergic nRT neurons were hypoexcitable compared to the control, firing fewer spikes during both tonic and burst firing.This contrast between the Ritter-Makinson and Studtmann studies on howScn1adeficits change nRT physiology is truly interesting.The major differences between the studies were transgenic mouse model and age range.That different models or ages could result in opposing changes to the physiology of a specific cell type, while still resulting in the same disease outcome, affirms the importance of in-depth study of disease models beyond initial phenotypes and intuitive hypotheses.
Finally, Mattis et al.(2022) examined cell excitability and circuit processing in another key locus of seizure activity in DS, the dentate gyrus.These experiments were performed inScn1a+/–mice at ages at which PV interneuron excitability was normalized to almost wild type levels.They found hyperactivity within this circuit, but due to increased excitatory synaptic input onto dentate gyrus granule cells.Importantly, this effect was still present with inhibition blocked, showing that it was not due to reduced inhibitory tone.They also showed that activation of abnormally stronger entorhinal cortical inputs to the dentate gyrus could trigger seizures, while at this developmental stage, PV interneurons were providing strong seizure control.While mechanisms by which these excitatory synapses have been strengthened in the DS model are unclear, these data provide another example of how unexpected changes to circuit interactions are causing abnormal activity despite normalized PV+interneuron physiology.
Overall, these recent studies show that the phenotypes underlying seizures and neuropsychiatric comorbidities of DEEs and DS are broader, more complex, and more subtle than initially understood.Importantly, in parallel to these physiological studies, genetic studies have been working to map out mechanisms underlying major differences in phenotypes between mouse model strains (e.g., Kearney et al., 2022), which will aid in our understanding of the broad differences in penetrance and severity of symptoms experienced by people with DEE gene variants.The evolution of this field of study from the straightforwardSCN1A/interneuron hypothesis of DS to a hypothesis inclusive of multiple cell types, brain regions, modifier genes, and direct and indirect changes serves as an example of the importance of rigorous and creative experimental design to test new questions rather than letting an intuitive initial hypothesis become the dogma of the field.The author would like to acknowledge the work of members of the Howard Lab, in particular Jessica
Chancey, PhD, for their hard work and insights, as well as Audrey Brumback, MD, PhD, and members of the Brumback Lab for their support.The author also wishes to thank the Dravet Syndrome Foundation and members of the epilepsy research community for ongoing support and collegiality.
This work was supported by Dravet Syndrome Foundation, NIH/NINDS R01-NS112500.
MacKenzie A.Howard*
Department of Neurology, Dell Medical School,Austin, TX, USA
Center for Learning and Memory and Department of Neuroscience, University of Texas at Austin,Austin, TX, USA
*Correspondence to: MacKenzie A.Howard, PhD,mackenziehoward@austin.utexas.edu.https://orcid.org/0000-0003-2832-6873(MacKenzie A.Howard)
Date of submission: June 19, 2023
Date of decision: October 30, 2023
Date of acceptance: November 9, 2023
Date of web publication: December 15, 2023
https://doi.org/10.4103/1673-5374.390967 How to cite this article:Howard MA (2024) New complex physiological findings evolve hypothesized mechanisms of Dravet syndrome.Neural Regen Res 19(9):1867-1868.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- NADPH oxidase 4 (NOX4) as a biomarker and therapeutic target in neurodegenerative diseases
- Circadian rhythm disruption and retinal dysfunction:a bidirectional link in Alzheimer’s disease?
- Interplay between the glymphatic system and neurotoxic proteins in Parkinson’s disease and related disorders: current knowledge and future directions
- Roles of neuronal lysosomes in the etiology of Parkinson’s disease
- Therapeutic advances in neural regeneration for Huntington’s disease
- The advantages of multi-level omics research on stem cell-based therapies for ischemic stroke