
1个北方汉族家族性免疫球蛋白A肾病的家系分析和致病基因诊断
2024-01-10 10:55杨媛茹李增艳
内科 2023年6期
杨媛茹 李增艳
内蒙古科技大学包头医学院第一附属医院肾内科,包头市 014000
免疫球蛋白A(immunoglobulin A,IgA)肾病是一种最常见的原发性肾小球疾病,通常伴随着不同程度的蛋白尿、无症状血尿和渐进性的肾功能损伤。相关资料显示[1-2],中国人群患IgA肾病占全部原发性肾小球疾病的30%~45%,20%~40%的IgA肾病患者在确诊后的20年内会发展成终末期肾病。家族性IgA肾病是IgA肾病的分型之一,以X连锁隐性或常染色体显性方式遗传[3-4]。由于对家族性IgA肾病的患病率和临床特点的了解不全面,故临床诊治的IgA肾病患者里可能包含家族性IgA肾病患者,导致漏诊,并延误家族中其他未发病的家庭成员的早期诊断和治疗。20世纪70年代末Tolkoff-Rubin等[5-6]报告了2个家族性IgA肾病家系后,越来越多的学者报告家族性IgA肾病家系[7-18],这一疾病的家族易感性得到关注。INF2蛋白属于成蛋白家族,是一种重要的肌动蛋白成核因子,可促进肌动蛋白的聚合和解聚,维持细胞骨架的正常结构与功能[19]。既往文献报告[20-21],INF2基因突变可导致家族性蛋白尿疾病。本研究分析2002年发现的1个北方汉族家族性IgA肾病家系的临床资料,对其致病基因进行定位研究,旨为后续家族性IgA肾病的研究打下基础。
1 资料与方法
1.1 研究对象 选择我院肾内科于2002年发现的1个北方汉族家族性IgA肾病家系,5代共计26名成员。该家系成员均对本研究知情同意。
1.2 一般资料的收集 采集该家族成员的一般资料,包括年龄、性别、婚育情况、身体状况、现患病的疾病进展程度、肾功能分级、肾脏病理活检分型,以及去世亲属的存活年龄、死因和存活期身体状况,并构建家系图。
1.3 临床资料的收集 采集该家系现存成员的外周静脉血、随机尿液和24 h尿液,并及时冷藏保存;常规检测肾功能、尿常规、尿微量白蛋白、尿红细胞位相、24小时尿蛋白定量等。
1.4 致病基因的检测 将外周静脉血样本送至北京大学第一医院,应用全外显子组测序行基因检测,探究是否存在与疾病表型相关的致病性基因突变。
1.5 疾病诊断 查阅国内外文献,结合该家系成员的临床资料和基因测序结果进行诊断。
1.6 并发症的预测 应用REVEL蛋白功能预测软件分析基因突变位点对蛋白质结构和功能的影响,并考虑下一步可能出现的并发症。
2 结 果
2.1 家系图谱 该家系5代共计26名成员,详见图1。
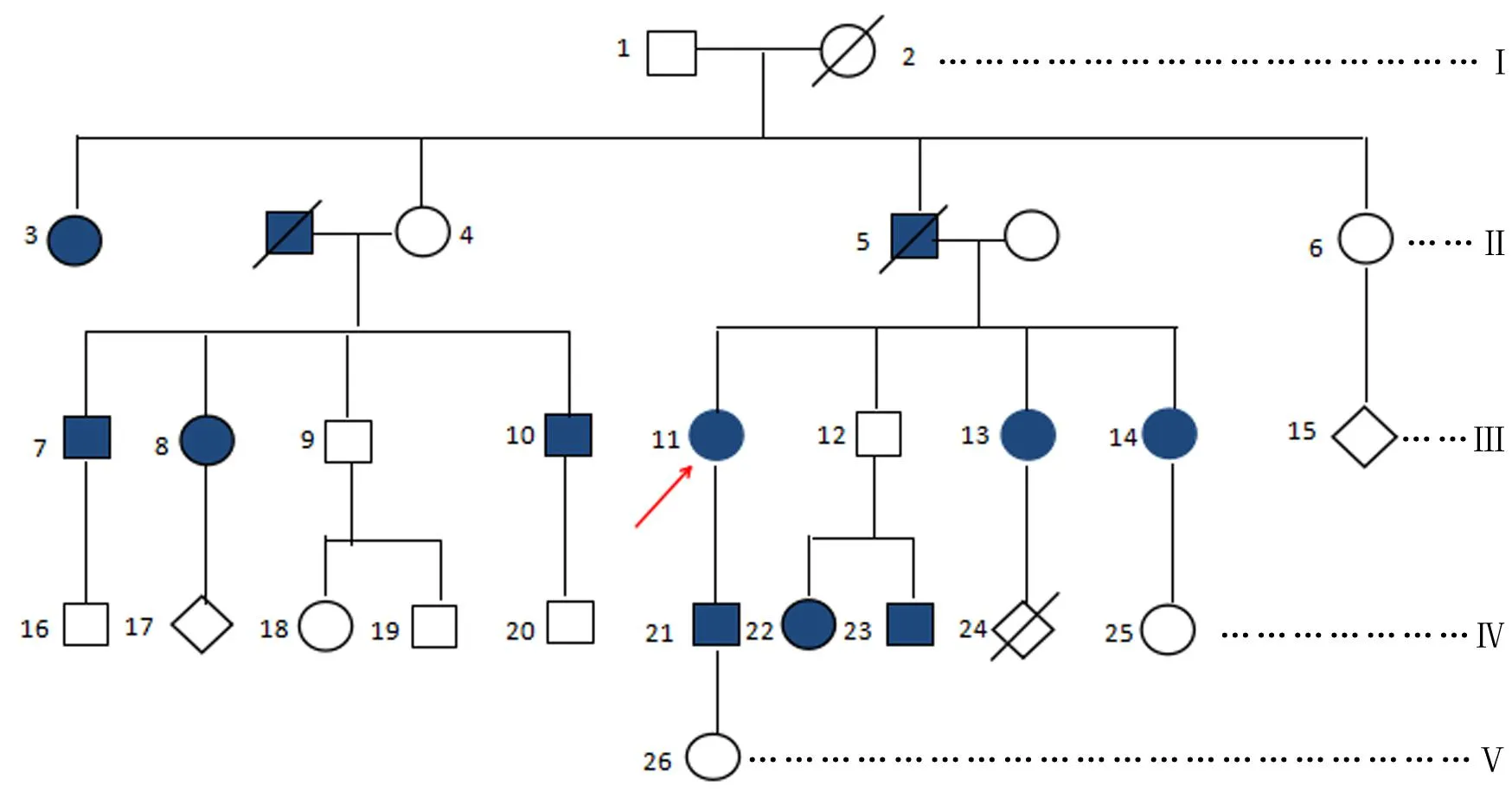
图1 家族性IgA肾病的家系图谱
2.2 家族成员的临床资料 先证者主诉以“发现蛋白尿13年”入院,伴腰痛、周身乏力、纳差、尿量减少,尿常规检测蛋白尿2+、血尿1+,自身免疫抗体、免疫球蛋白补体、感染性疾病筛查和血液系统肿瘤等相关指标均为阴性,泌尿系彩超和肾功能化验提示肾功能低下。该家系共计12名成员有不同程度的肾脏受累,6名成员临床表现为蛋白尿,3名成员临床表现为镜下血尿,先证者、家系13号和21号肾脏病理活检诊断为系膜增生性IgA肾病。
2.3 基因检测 全外显子组测序检出INF2基因c.653G>A(p.R218Q)的杂合突变。REVEL蛋白功能预测软件分析INF2基因的c.653G>A(p.R218Q)突变属于致病突变,可引起局灶性节段性肾小球硬化症,详见图2。

图2 REVEL蛋白功能预测软件分析INF2基因c.653G>A(p.R218Q)突变结果
2.4 该家系成员符合家族性IgA肾病 该家系具有以下特点:(1)连续3代有发病者,男女均有发病,符合常染色体显性遗传的特征;(2)3名家系成员肾脏病理活检诊断为系膜增生性IgA肾病;(3)家系成员的临床检查和肾脏病理活检排除了高血压肾病、糖尿病肾病等继发性肾病;(4)以该家系成员发病的年龄来看,第2代的家庭成员约在老年时期发病,但第3、第4代家系成员的发病年龄显著提早(遗传早现);(5)该家系成员的疾病严重程度和疾病预期治疗效果不佳,共有12名成员发病,其中有2名成员因慢性肾衰竭5期死亡,有1名成员已做肾脏移植手术,其余的9名成员均有持续多年的不同程度的镜下蛋白尿或血尿或血清尿酸升高或肾功能异常。根据目前的诊断标准[3,12-14],可明确诊断该家系存在家族性IgA肾病。
3 讨 论
在本研究中,全外显子组测序检出INF2基因c.653G>A(p.R218Q)的杂合突变,其机制是653号的核苷酸由鸟嘌呤G突变为腺嘌呤A(c.653G>A),导致第218号氨基酸由精氨酸变为谷氨酰胺(p.R218Q)。根据美国医学遗传学与基因组学学会指南,该变异初步判定为致病性变异PS4+PM1+PM2+PM5+PP3。INF2基因突变可导致遗传性运动感觉性周围神经病(OMIM:614455)和局灶性节段性肾小球硬化症(OMIM:613237)。遗传性运动感觉性周围神经病是常染色体显性遗传病,患者一旦发病都具有蛋白尿表现(伴或不伴显微血尿),有些患者伴有肾病综合征。局灶性节段性肾小球硬化症患者的临床特征为肾小球内节段性硬化、蛋白尿、肾小球滤过率降低和肾功能进行性下降,肾功能不全常常发展为终末期肾病,严重时需要透析治疗或肾脏移植。
家族性IgA肾病是IgA肾病的特殊类型,以X连锁隐性或常染色体显性方式遗传[3-4],其临床表现与散发性IgA肾病相似,具有发病率高、预后差和遗传早现等特点[13]。根据资料统计[9],意大利南部地区大约有一半以上的IgA肾病呈家族聚集性发病;北京大学肾脏病研究所的王海燕教授曾对1988年至2001年间确诊的777名IgA肾病患者进行分析,发现有8.7%的患者呈家族聚集性发病[16]。由于IgA肾病的发病年龄和起病程度不同,在临床诊治的过程中,医生很容易忽略患者是否有肾病家族史,因此在临床诊治的IgA肾病中可能涵盖了家族性IgA肾病。同时,相较于散发性IgA肾病,家族性IgA肾病的患者发病时病情往往不重[22],但病程后期常发展为终末期肾病,预后差。
综上所述,及早发现、及早诊断家族性IgA肾病是至关重要的。因此,临床医生对于肾脏病理活检证实为IgA肾病的患者或者有肾功能异常的疑似患者,应详细询问其家族史并进行家族成员的尿检筛查,强调普及基因测序的重要性,使更多的家族性IgA肾病家系通过基因测序确定其家系的致病基因,早期、准确确定其诊断,并准确地鉴别诊断,及时调整治疗方向并进行精细化基因治疗,最大程度地延缓肾脏功能的恶化,减慢疾病的发展进程,最大限度地延长生命周期,使这些患者能够在后续的临床诊疗中得到更好的指导和治疗,更加准确地判断预后。
本研究中的中国北方汉族家族性IgA肾病家系相对完整,患病家庭成员和健康家庭成员的比例也较为适中,有利于接下来对致病基因的连锁分析和突变位点对蛋白结构和功能影响的分析[15],也为研究中国人群的家族性IgA肾病家系提供一定的临床资料。
猜你喜欢
昆明医科大学学报(2021年12期)2021-12-30
中国民间疗法(2021年19期)2021-11-20
中国民间疗法(2021年18期)2021-11-02
中国卫生标准管理(2015年25期)2016-01-14
中国卫生标准管理(2015年25期)2016-01-14
广东海洋大学学报(2015年4期)2016-01-13
中国继续医学教育(2015年2期)2016-01-06
听力学及言语疾病杂志(2015年5期)2015-12-24
首都医科大学学报(2015年4期)2015-12-16
中国当代医药(2015年22期)2015-03-01
