
方解石晶体电子结构及(104)面表面特性第一性原理计算
2024-01-08 06:44谢瑞琦赵志辉周武昌
金属矿山 2023年12期
谢瑞琦 王 勋 赵志辉 周武昌
(1.矿冶科技集团有限公司矿物加工科学与技术国家重点实验室,北京 100160;2.昆明理工大学国土资源工程学院,云南 昆明 650093)
方解石是一种碳酸盐矿物,是最常见的天然碳酸钙,分布极广,常与钨矿、萤石、菱镁矿、锡石等矿物共伴生,是常见的脉石矿物之一。由于与方解石共伴生的目的矿物通常含有Ca、Mg等与方解石表面活性位点活性相近的位点或Sn等较易形成螯合作用的位点,导致方解石与目的矿物分离困难[1-4]。
为了更有效地将方解石与目的矿物分离,研发高选择性的浮选药剂具有重要意义,而浮选药剂的选择性与矿物表面特性息息相关。第一性原理计算为研究者从微观角度探明矿物晶体表面特性提供了良好的渠道。目前关于方解石结构的第一性原理计算研究主要分为以下几类,第一类是研究方解石与水分子间的作用,如王杰等[5]利用第一性原理研究了方解石的晶体结构及其与水分子及水分子簇的吸附作用,研究结果表明方解石在与水反应时O的活性最强,其Ca和O位点与单个水分子形成吸附,水分子簇与方解石作用时,水分子间及水分子与方解石表面均存在氢键作用。第二类是研究某一浮选药剂与方解石间的作用,如张多阳等[6]计算了菱镁矿、白云石和方解石的能带、态密度及与常见捕收剂、抑制剂作用,为矿物浮选分离药剂的选择提供了参考。张英等[7]计算了白钨矿、萤石和方解石的电子结构及聚丙烯酸钠与矿物作用的吸附能。第三类是研究金属掺杂对方解石的影响,如陈鸿[8]研究了铁、镁、铝等元素对方解石和白云石晶体结构和表面性质的影响,结果表明,镁、铝、铁在常温下不易在方解石中通过晶格取代形成杂质缺陷,铁杂质会降低水分子在方解石表面的吸附。
以上研究阐明了方解石部分表面特性及其与常见药剂的作用,但未对方解石体相的特性及切表面后的变化、表面电荷差分密度及与常见极性基的作用进行系统研究。本文利用Materials Studio软件计算分析了方解石晶体体相和方解石(104)面的布居、能带及总态密度和分态密度,并计算分析了方解石(104)面的弛豫、电荷差分密度以及与常见极性基的前线轨道能级差。通过从微观角度分析方解石的表面特性,本文揭示了方解石体相晶体特征、方解石(104)面表面活性位点的电子特性及其与药剂极性基间的作用,可为研发高选择性方解石浮选药剂提供参考。
1 模拟与计算方法
1.1 方解石晶体结构及优化
方解石的晶体结构原始数据来源于American Mineralogist Crystal Structure Database[9]。采用Materials Studio中的CASTEP模块对方解石结构进行优化。优化过程中,参考文献[10-11],交换相关泛函为GGA-PBE、k点取样密度为4×4×2、截断能设置为500 eV。赝势采用OTFG超软赝势。自洽迭代过程中的收敛标准为:能量最大变化不超过2×10-5eV,原子最大位移不超过0.000 2 nm,原子间内应力不超过0.1 GPa,SFC收敛标准为1×10-6eV。最终优化得到的方解石晶胞参数为a=b=0.499 0 nm,c=1.706 1 nm,α=β=90.00°,γ=120.00°,与文献[5]中的计算结果及实验值均接近,说明构建的晶体结构合理,具有代表性。
1.2 表面模型建立
方解石(104)面为方解石常见解理面,因此选择方解石(104)面作为研究对象。在尽可能缩短计算时间且能维持稳定的表面性能及尽量避免上下两底面间的干扰的情况下,构建了切割厚度为3,真空层厚度为2.0 nm的方解石(104)表面slab模型。优化后的方解石晶胞及优化后的方解石(104)表面如图1所示。
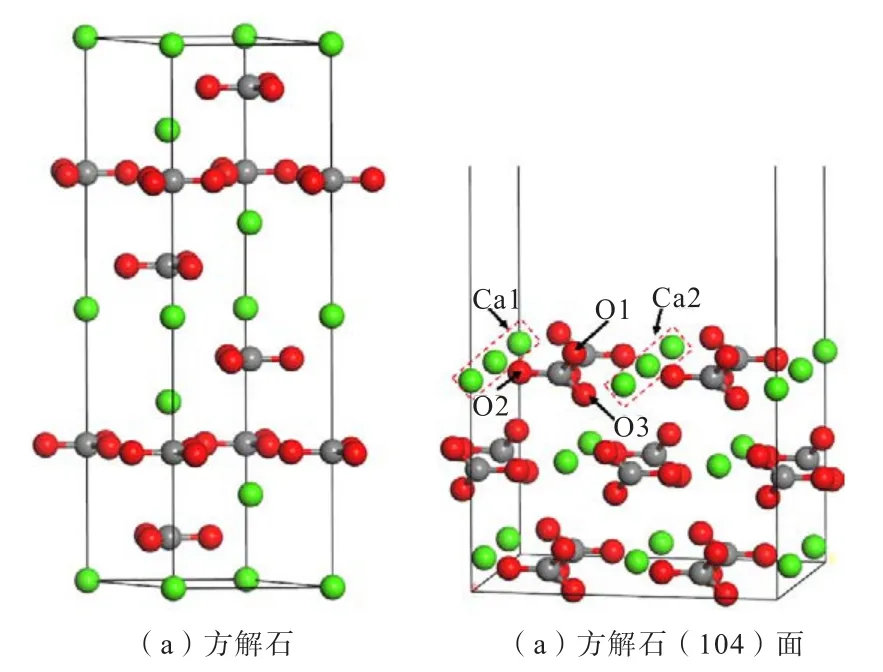
图1 优化后的方解石晶胞和优化后的方解石(104)面Fig.1 The optimized calcite cell and optimized calcite (104) surface
2 计算结果与讨论
2.1 方解石晶体分析
2.1.1 布居分析
布居为原子核外电子的分布排列位置,方解石的原子Mulliken布居结果如表1所示。

表1 方解石原子Mulliken布居Table 1 The atom Mulliken population of calcite
从表1可以看出:方解石同种元素化学环境一致,说明方解石结构对称性较强。方解石中荷正电的原子为C和Ca,其中C所带电荷为0.79 e,其价电子构型为C 2s0.842p2.37,s轨道失去了1.16个单位的电子,p轨道得到了0.37个单位的电子,主要是3 s轨道失去电子;Ca所带电荷为1.40 e,其价电子构型为Ca 3p64s2.11,s轨道得到了0.11个单位的电子。方解石中荷负电的原子为O,其所带电荷为-0.73 e,其价电子构型为O 2s1.802p4.93,s轨道失去了0.20个单位的电子,p轨道得到0.93个单位的电子,主要是p轨道得到电子。
矿物Mulliken键布居可反映化学键的性质,其值越接近0,表明键的离子性越强,而其值越接近1,表明键的共价性越强[12]。方解石的Mulliken键布居值如表2所示。

表2 方解石Mulliken键布居Table 2 The bond Mulliken population of calcite
由表2可知,方解石中的Ca-O和C-O布居值仅有1种,说明方解石对称性较强,结构简单[13]。Ca-O键的布居值为0.11,说明其具有一定的离子性,C-O键的布居值为0.83,说明其具有较强的共价性,Ca-O的键长远大于C-O的键长。破碎磨矿时,Ca-O键最易断裂,因此,Ca为方解石表面的阳离子活性位点,O为方解石表面的阴离子活性位点。
2.1.2 能带和态密度分析
方解石的能带间隙宽度为5.101 eV,因此为绝缘体[12]。图2所示是方解石总态密度图和各原子的分态密度图。方解石的总态密度主要分为4部分;第1部分为-39~-36 eV之间,这部分完全由Ca的s轨道组成;第2部分为-24~-15 eV之间,这部分Ca、O、C均有贡献,其中,C主要由s轨道贡献低能部分,p轨道和少量s轨道贡献高能部分;O主要由s轨道贡献,p轨道贡献较小;Ca主要由p轨道和极少量d轨道贡献;第3部分为-9~0 eV之间,该部分主要由O和C及少量的Ca参与构成。其中,C的s轨道和p轨道主要组成-9~-3 eV的低能部分;O的s轨道组成低能部分,p轨道在整个能量范围内都有较大的贡献,特别是在费米能级附近,因此O是方解石中最活跃的原子[14]。此外,Ca在费米能级附近也有少量的贡献,说明Ca也有一定的活性;第4部分为3~18 eV之间,所有原子都有轨道参与贡献。

图2 方解石总态密度图和各原子分态密度图Fig.2 The total and partial density of state of calcite
2.2 方解石(104)表面特性
2.2.1 方解石(104)面的结构弛豫
方解石晶体中原子呈周期性分布,但切表面后,表面原子将不再呈现出周期性分布,表面原子的受力也将发生改变,为了使系统平衡,表面原子会通过弛豫而发生重构现象[15]。方解石(104)面表面原子优化前后的分数坐标变化值如表3所示。
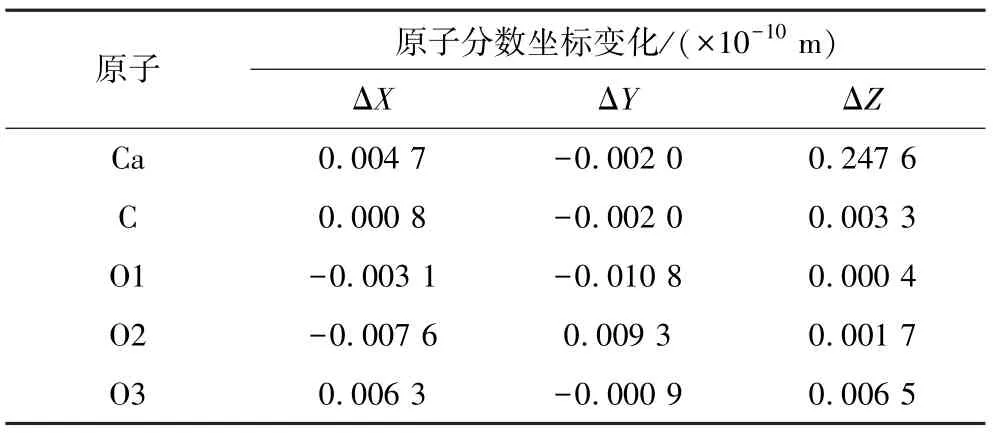
表3 方解石(104)面表面原子优化前后的分数坐标变化值Table 3 The change of fractional coordinate value of atoms on calcite (104) surface before and after the optimization
由表3可知,方解石的表面原子在优化以后,均产生了一定弛豫现象。Ca原子在Z轴方向的弛豫最为明显,C在3个方向的弛豫相对较小,碳酸根的3个O原子在3个方向表现出了不同的弛豫程度。
2.2.2 方解石(104)面的布居分析
方解石(104)面表面原子之间的相对位移以及配位数相较于体相发生了变化,原子外层电子和键长都随之发生变化。表4为方解石(104)面表面原子的Mulliken布居分布,可以看出相较于方解石体相结构,方解石(104)面表面原子的外层电子都发生了一定程度的变化。C的价电子构型变为C 2s0.832p2.36,s轨道相较于体相多失去了0.01个单位的电子,p轨道相较于体相少得到了0.01个单位的电子,总电子数减少为3.18,电荷变为0.82 e。O的价电子构型变为2种,分别为O 2s1.802p4.91和O 2s1.812p4.96,相较于体相,其s轨道分别少失去了0个单位的电子和0.01个单位的电子,p轨道分别多失去了0.02个单位的电子和少失去了0.03个单位的电子,所带电荷分别为-0.72 e和-0.77 e。Ca的价电子构型变为Ca 3p64s2.10,与体相变化不大,s轨道相较于体相少得到了0.01个单位的电子,p轨道无变化。虽然Ca仅具有一种价电子构型,但表面存在具有2种电子总数的Ca,电子总数分别为8.56和8.55,电荷分别为1.44 e和1.45 e。

表4 方解石(104)面表面原子的Mulliken布居Table 4 The atom Mulliken population of calcite (104) surface
表5为方解石(104)面表面电子的键布居。由表5可知,方解石表面的Ca-O键和C-O键的布居值和键长较体相发生了明显改变,其对称性变差。C-O键的布居值由体相的0.83变为0.89和0.78。Ca-O键的布居由体相的1种键布居值0.11变为3种键布居值0.14、0.11和0.10。此外,当Ca-O布居值为0.11时,其键长较体相的0.235 4 nm变长,键强变弱。

表5 方解石(104)面表面原子的Mulliken键布居Table 5 The bond Mulliken population of calcite (104) surface
2.2.3 方解石(104)面的态密度分析
图3为方解石(104)面表面原子的总态密度图和各原子的分态密度图。与体相类似,方解石(104)面的总态密度也分为4部分,且能量范围基本一致,但态密度发生了变化。第1部分为-39~-36 eV之间,这部分完全由Ca的s轨道组成;第2部分为-24~-15 eV之间,这部分Ca、O、C均有贡献,其中,Ca主要由p轨道,C主要是s轨道贡献,O的s轨道和p轨道均有较大贡献;第3部分为-9~0 eV之间,这部分主要由C和O的s轨道和p轨道构成,少量Ca参与贡献,第4部分为3~18 eV之间,所有原子都有轨道参与贡献。与体相一致,费米能级附近主要由O的p轨道和少量Ca的d轨道构成,因此,O为方解石(104)面最活跃的原子,其次是Ca原子。
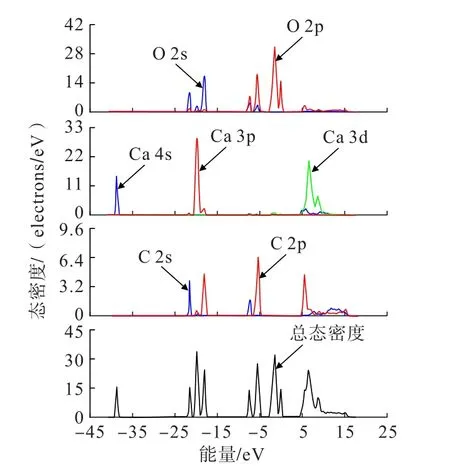
图3 方解石(104)面表面各原子总态密度图和分态密度图Fig.3 The total and partial density of state of calcite (104) surface
2.2.4 方解石(104)面的差分电荷密度图
方解石(104)面表面原子的差分电荷密度如图4所示,深色区域表明电荷密度高,浅色区域表明电荷密度低。Ca-O键电荷密度较C-O键电荷密度低,说明Ca-O键的键能较C-O键低,这与布居分析结果一致。此外,方解石(104)面中O为多电子位点,C和Ca为缺电子位点,缺电子位点和多电子位点交替出现,得失电子程度不同,各原子表现出不同的化学活性。

图4 方解石(104)面表面差分电荷密度Fig.4 The electron density difference of calcite (104) surface
2.2.5 方解石(104)面前线轨道能级差
物质发生化学反应时,物质HOMO中的电子分别流向对方的LUMO,只有物质A(或B)的HOMO与物质B(或A)的LUMO的能量比较接近,对称性也互相匹配时,才容易发生电子转移,当物质A的HOMO与物质B的LUMO能级差和物质B的HOMO与物质A的LUMO能级差不一致时,电子转移以能级差绝对值低的流向为主。由于阳离子捕收剂主要以静电吸附和氢键作用与矿物表面作用,而阴离子捕收剂主要以键合吸附为主,因此,本部分主要计算常见阴离子捕收剂的极性基与方解石间的HOMO和LUMO的最低能级差绝对值|ΔE|min来预测化学相互作用的难易程度。|ΔE|min越小,说明电子转移越容易发生,即反应越容易发生[16-18]。表6所示是极性基团与方解石(110)面的前线轨道能级差。

表6 阴离子基团与方解石(104)面表面的前线轨道能量Table 6 The frontier molecular orbital energy of anionic group and calcite (104) surfaces eV
由表6可知,-COOH、-CONHOH、-OSO3H等基团[19-21]与方解石(104)面间的能级差绝对值相对较小,说明方解石(104)面易与阴离子捕收剂作用,其中,-OSO-3最易与方解石(104)面作用。方解石(104)面表面荷正电的活性位点为Ca,其易与含-COOH、-CONHOH、-OSO3H等官能团的阴离子捕收剂作用,其中,含-OSO-3基团的药剂可能最易与方解石(104)面作用。
3 结 论
(1)方解石晶体中,荷正电的原子为Ca和C,荷负电的原子为O。方解石同种元素化学环境一致,说明方解石结构对称性较强,结构简单。Ca-O键具有较强的离子性,C-O键具有较强的共价性,因此,Ca-O键最易断裂,Ca为方解石表面的阳离子活性位点,O为阴离子活性位点。
(2)方解石的能带间隙宽度为5.101 eV,为绝缘体。其能带主要分为4个部分,O和Ca在费米能级附近的态密度均有贡献,因此O和Ca是方解石中的活性原子,其中O的p轨道在费米能级附近的态密度贡献最大,因此O是方解石中最活跃的原子。
(3)方解石(104)面表面原子价电子构型、布居、键长、态密度等较体相均发生了变化,但费米能级附近主要还是由O的p轨道和少量Ca的d轨道构成。差分电荷密度图表明方解石(104)面Ca-O键电荷密度较C-O键电荷密度低,说明Ca-O键的键能较C-O键低,不同种类原子表现出不同的电子得失情况。
(4)极性基团与方解石(110)面的前线轨道能级差表明:方解石(104)面易与含-COOH、-CONHOH、-OSO3H等官能团的阴离子捕收剂作用,其中,含-OSO-3基团的药剂可能最易与方解石(104)面作用。
猜你喜欢
硅酸盐通报(2022年8期)2022-09-08
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学生数理化·中考版(2021年10期)2021-11-22
矿产综合利用(2021年3期)2021-07-14
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
矿产综合利用(2020年1期)2020-07-24
新高考·高一物理(2015年6期)2015-09-28
新高考·高一物理(2015年6期)2015-09-28
