
Autophagy modulates physiologic and adaptive response in the liver☆
2023-12-29 01:21TrinhVanLeNhungHaiTruongAiXuanHolterman
Liver Research 2023年4期
Trinh Van Le ,Nhung Hai Truong ,Ai Xuan L.Holterman
a Laboratory of Stem Cell Research and Application, University of Science-VNUHCM, Ho Chi Minh City, Vietnam
b Vietnam National University, Ho Chi Minh City, Vietnam
c Faculty of Biology and Biotechnology, University of Science-VNUHCM, Ho Chi Minh City, Vietnam
d Department of Pediatrics and Surgery, University of Illinois College of Medicine, Chicago and Peoria, IL, USA
Keywords: Autophagy Biliary epithelial cell Hepatitis Hepatocellular carcinoma Hepatic stellate cell Hepatocyte
ABSTRACT Autophagy is a physiological process that is ubiquitous and essential to the disposal or recycling of damaged cellular organelles and misfolded proteins to maintain organ homeostasis and survival.Its importance in the regulation of liver function in normal and pathological conditions is increasingly recognized.This review summarizes how autophagy regulates epithelial cell-and non-epithelial cellspecific function in the liver and how it differentially participates in hepatic homeostasis,hepatic injury response to stress-induced liver damage such as cholestasis,sepsis,non-alcoholic and alcohol-associated liver disease,viral hepatitis,hepatic fibrosis,hepatocellular and cholangiocellular carcinoma,and aging.Autophagy-based interventional studies for liver diseases that are currently registered in clinicatrials.gov are summarized.Given the broad and multidirectional autophagy response in the liver,a more refined understanding of the liver cell-specific autophagy activities in a context-dependent manner is necessary.
1.Introduction
Autophagy is a conserved biological process in eukaryotic organisms for cellular housekeeping;as a physiological response to nutrients,internal or external stimuli;and during reactive oxygen species (ROS) oxidative and endoplasmic reticulum (ER) stress reaction to hypoxia,infection,metabolic dysfunction,etc.During autophagy,damaged organelles and large aggregated misfolded proteins are eliminated or recycled through the lysosomal compartment during the unfolded protein response (UPR) as a synergistic pathway to the ubiquitin-proteasome system (UPS)route of degradation and recycling for short-lived proteins.Since the liver is an essential organ for lipid,glucose,and amino acid metabolism,as well as for endobiotics and xenobiotics disposal,a well-coordinated autophagy response is essential for normal liver function,while disruption of such a response initiates and exacerbates hepatic injury.1-4
This manuscript will characterize the general autophagy process in the liver physiological response and the liver-specific autophagy adaptation to nutrients,abnormal fat,and bile acid handling;to hepatic inflammation and fibrosis;from cell proliferation,differentiation,and dedifferentiation to carcinogenesis.The review will focus on thein vitroautophagy response of specific cellular compartments in the liver that are most critical to hepatic function (i.e.the liver epithelial cells (hepatocytes and biliary epithelial cells (BECs),also referred to as cholangiocytes) and the non-epithelial cells (hepatic stellate cells (HSCs) and monocytes such as Kupffer cells) to ROS,oxidative,mitochondrial,lysosomal and ER stress;examine theirin vivohepatic effects in the context of commonly seen clinical entities such as fatty liver;pathological conditions with underlying inflammatory characteristics from non-alcoholic steatohepatis,bile acid-mediated cholestasis,sepsis to alcohol-associated,and viral hepatitis;those secondary to chronic inflammation as major contributing factor such as hepatic fibrosis,cirrhosis,and hepatocellular carcinoma;with a brief overview of the role of autophagy in liver senescence and currently registered clinical trials of autophagy-based interventions in liver diseases.
2.Autophagy description
The three main types of autophagy,macroautophagy,chaperonemediated autophagy (CMA),and microautophagy,are defined by how the contents are delivered to the lysosomes for lysosomal hydroxylase digestion(See Fig.1).
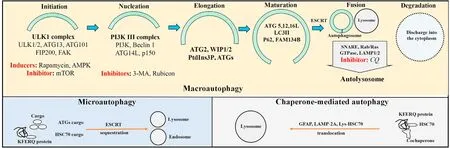
Fig.1.Macroautophagy,chaperone-mediated autophagy,and microautophagy.Macroautophagy involves a multistep processing of cargo involving Atg complexes for initiation,nucleation,elongation,and maturation for double-membrane autophagosome formation and fusion with lysosomes to generate autolysosome.Inducers of macroautophagy include AMPK,rapamycin which suppresses mTOR autophagy inhibitors.Inhibitors of macroautophagy are mTOR,3-MA,rubicon,and CQ.Microautophagy involves cytoplasmic cargo-ATG protein/adaptor complex for direct delivery to lysosomes,or of the KFERQ motif-bearing proteins -Hsc70/90 -ESCRT complex for endocytosis-like engulfment by the lysosome.Chaperone-mediated autophagy recruits a translocation system of channel-like proteins such as GFAP,LAMP-2A,and Lys-HSC70 for cargoes containing the KFERQ motif-bearing proteins-Hsc-70/90 complex.Abbreviations: Atg,autophagy-related gene;Hsc70,70 kDa heat shock protein;LAMP-2A,lysosome-associated membrane protein-2A;Lys-HSC70,lysosomal hsc70;GFAP,glial fibrillary acidic protein;AMPK,adenosine 5′-monophosphate activated protein kinase;mTOR,mammalian target of rapamycin;3-MA,3-methyladenine;CQ,chloroquine.
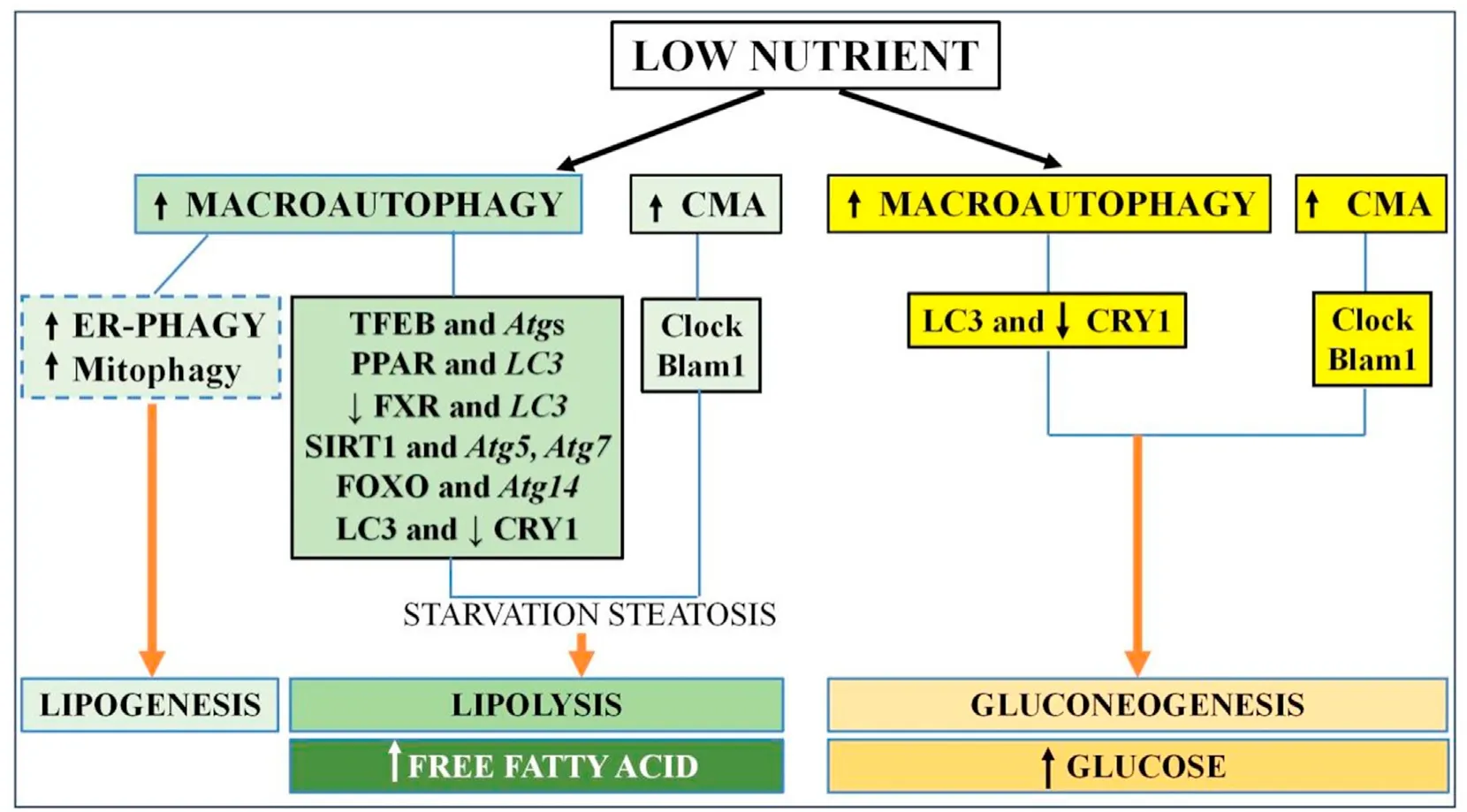
Fig.2.The liver autophagy response to starvation.Macroautophagy and CMA are upregulated in the low-nutrient state with two different outcomes,lipolysis and gluconeogenesis.CMA function is synchronized with hepatic CLOCK genes(illustrated as Clock,Blam1,etc)and macroautophagy is modulated by autophagy transcriptional regulators TFEB,SIRT1,FoxO1,and PPARα with their corresponding target genes Atgs (such as Atg5, Atg7, LC3) to generate free fatty acid or glucose as energy substrate.Note that ER-phagy and mitophagy are reported to lead to lipogenesis.Abbreviations: CMA,chaperone-mediated autophagy;CRY1,cryptochrome 1;TFEB,transcription factor EB;FoxO1,forkhead box protein O1;PPARα,peroxisome proliferator-activated receptor alpha;Atg,autophagy-related gene;LC3,light chain 3;FXR,farnesoid-X receptor;ER,endoplasmic reticulum;SIRT,sirtuin.
Macroautophagy is the best-characterized form of autophagy.In macroautophagy,the cytoplasmic cargoes are transported insidede novodouble-membrane autophagosome vesicles that become auto(phago)lysosome after their fusion with the lysosomal membranes.5Macroautophagy is a complex stepwise process,from phagophore initiation,nucleation,elongation,and maturation into phagosome;autolysosome formation by fusion of the autophagosome with the endosome/lysosome;and lysosomal degradation of the autophagosome and its contents (Fig.1).The multiple regulatory proteins that carry out the macroautophagy steps include the core autophagy-related genes (Atg) protein family.Briefly,the process consists chronologically of:(1)The stable ULK kinase UNC-51-like kinase 1/2 (ULK1/2) -ATG13-focal adhesion kinase (FAK)-focal adhesion kinase family interacting protein of 200 kD(FIP200)-ATG101 complex which responds to autophagy inducing signals to initiate phagophore formation.6(2) Phagophore nucleation by recruitment of the class III phosphatidylinositide 3-kinase(PI3K III)complex which consists of Class III PI3K(also referred to as vacuolar protein sorting 34 (VPS34))-Beclin1 (or Atg6)-p150-ATG14L to the ULK complex.(3) Phagophore expansion by recruitment of the ATG2-wild-type p53-induced phosphatase 1/2 (WIP1/2) complex to the PI3K complex for phosphorylation of the phosphatidylinositol moiety to form phosphatidylinositol 3-phosphate (PtdIns3P),and recruitment of additional ATG proteins to PtdIns3P.(4) Phagophore elongation and maturation by further association with ubiquitin-like ATG12-ATG5-ATG16L and with microtubuleassociated protein light chain 3 (LC3) II by conjugation of LC3I with phosphatidylethanolamine (PE).7(5) Phagophore membrane maturation and closure to form autophagosome involving endosomal sorting complex required for transport (ESCRT).8(6) Autophagosome and lysosome fusion to form autophagolysosome,involving soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins,multiple small Ras-related proteins in brain (Rab)/Ras GTPases,and lysosomal associated membrane protein 1(LAMP 1)and LAMP 2 lysosomal proteins;and(7) Lysosomal degradation of the cargoes,with release of the digested materials into the cytoplasm by various exporters in the lysosome membrane.6-10An essential step during autophagosome formation is the interaction of adaptor receptor proteins (such as sequestosome 1/p62 (SQSTM1/p62) for ubiquinated proteins and organelles,or protein family with sequence similarity 134 member B (FAM134B) for ER misfolded proteins) to recognize the cargo contents and deliver them to the phagosomes by association with LC3II at the phagophore.
Macroautophagy consists of selective and non-selective autophagy.Non-selective autophagy is responsive to nutrient fluctuation through nutrient sensor kinases such as autophagy inducer adenosine monophosphate(AMP)-activated protein kinase(AMPK)during starvation,and autophagy inhibitor mammalian target of rapamycin-1 (mTOR1) in the fed state.Selective autophagy is defined by the nature of the organelles which require ubiquitylation for recognition by the corresponding receptor proteins for association with LC3II before lysosomal engulfment.11It includes macrolipophagy for lipids,xenophagy for pathogens,mitophagy for damaged mitochondria and drug metabolites,pexophagy for peroxisomes,aggrephagy for protein aggregates,or ER phagy for ER fragments,....Autophagy is also modulated by gastrointestinal hormones,neuroendocrine signals and growth factors which regulate the hepatic nutrient response.12-14
Inducers of macroautophagy include AMPK,which phosphorylates and activates ULK1;and rapamycin,which inhibits mTOR.6Inhibitors of autophagy include Run domain Beclin-1 interacting and cysteine-rich-containing(Rubicon)protein which is a Beclin-1 interacting protein that binds to the PI3K complex to impair autophagosome/lysosome fusion and endocytic trafficking;mTOR which competes with AMPK phosphorylation of ULK1 to prevent ULK1 activation in nutrient-rich states or during the stress response;PI3K inhibitor 3-methyl adenine(3-MA)which prevents autophagosome formation;and chloroquine (CQ) which inhibits autophagosome-lysosome fusion.7,15-17
In microautophagy,non-selective bulk proteins are targeted into lysosomes while selective small-size cytoplasmic proteins with KFERQ-like motif are recognized by chaperone proteins such as 70 kDa heat shock protein (Hsc70) and by additional autophagyrelated adaptors.They are sequestered in late endosomes using endosomal sorting complexes required for transport (ESCRT) for lysosomal internalization via endocytosis-like lysosomal membrane engulfment or invagination.
In CMA,cytosolic proteins bearing the KFERQ-like amino acid motif are recognized by Hsc70 and directly delivered to the lysosomes through a translocation system of channel-like proteins such as glial fibrillary acidic protein (GFAP),lysosome-associated membrane protein-2A (LAMP-2A) and lysosomal Hsc70 (Lys-HSC70).
Macroautophagy,CMA,and microautophagy have distinct yet overlapping and potential mutual compensatory functions,including the response to starvation with glycolysis,proteolysis,and lipolysis for energy substrate generation.18-20
3.Autophagy response in the liver
3.1. Autophagy and the liver response to nutrients
3.1.1.Hepatic circadian rhythm and fasting
The 24-hour circadian cycle controls hunger,and food intake and synchronizes the basal liver clock to regulate nutrient metabolism.21This circadian rhythm is under the control of the “clock”genes encoded by transcription factors brain and muscle Arnt-like protein 1 (BMAL1) and circadian locomotor output cycles kaput(CLOCK),CCAAT enhancer binding protein beta (C/EBPβ);nuclear receptor subfamily 1 group D member 1 (Nr1d1) and Per1b,Reverbα for period genes1/2/3(PER)and cryptochrome genes(CRY1/2),many of which are highly expressed in the liver;display diurnal expression patterns and regulate glucose and lipid metabolism.22-28Mice with liver-specific inactivation ofBmal1exhibit hypoglycemia despite fasting,and abnormal circulating lipoprotein profile.Rev-erbα depletion leads to fatty liver.26,28,29Thus,dysregulation of hepatic circadian protein impairs nutrient metabolism.
3.1.2.Hepatic food circadian rhythm and autophagy
In the liver,autophagy activities are strongly linked to the liver clock function in metabolism.BMAL1,CLOCK,Rev-erbα,and CRY1 are degraded through autophagy in mouse liver,with the loss ofAtg7leading to disruption in the CLOCK protein expression.28,30,31Autophagy gene expression is,in turn,regulated by the clock proteins such as nuclear hormone receptors Nrd1 and Per1b in fasting zebrafish liver,and autophagy proteins are induced in mouse liver in a circadian manner by BMAL1:CLOCK.23,27Autophagy and liver clock proteins interact with each other to modulate nutrient processing.CRY1 clock protein acts as a suppressor of gluconeogenesis to maintain blood glucose in a time-related circadian rhythm.Under low nutrient conditions that stimulate non-selective autophagy,the interactions between liver CRY1 and LC3 induce hepatocyte autophagosome degradation of CRY1,thus preventing hypoglycemia,while worsening hyperglycemia with high-fat feeding.Conversely,inducible loss ofAtg7in the liver is associated with increased hepatic CRY1 and hypoglycemia from inhibition of gluconeogenesis.31Forkhead box protein O1 (FoxO1)transcription factor controls the expression of genes critical to gluconeogenesis,hepatic lipid metabolism,and autophagy.32-34FoxO1 transcriptionally regulates the expression ofAtg14,and mice with conditional deletion ofFoxO1in the liver have impaired expression of autophagy genes.Atg14,as a target gene of FoxO1,is expressed in a circadian oscillatory pattern,which jointly with Bmal1 and clock proteins,can orchestrate time-restricted lipid metabolism in primary hepatocytes and mouse liver.34
The circadian clock is also regulated by CMA which selectively targets Blam1 and Clock in mice liver;with disruption of CMA being linked to the accumulation of clock-dependent transcription and dysregulation of the circadian clock protein rhythm.35
3.1.3.Liver autophagy and normal nutrient response(See Fig.2)
3.1.3.1.Initial starvation steatosis autophagy response.With respect to physiologic autophagy-nutrient response,starvation induces hepatocyte autophagy to generate energy substrates through glucose metabolism,gluconeogenesis,and lipolysis.28,36-40Following intestinal digestion,triglycerides (TGs) and cholesterol esters are packaged in circulating lipid droplets(LDs)to be taken up and stored in hepatocytes.In conditions of amino acid deprivation wherede novofatty acid (FA) synthesis is inhibited,lipophagy is quickly induced in hepatocyte cell lines with accumulation of intracellular LD,leading to what is referred to as starvation steatosis.This is suppressed upon amino acid supplementation.41-43In vivo,mice with liver-specificAtg5defect indeed are resistant to starvation steatosis independently of underlyingde novolipogenesis or lipolysis.44Similarly,mice with liver-specific deletion of Sirtuin(SIRT),an autophagy regulator ofAtg5orAtg7,and mice with deletion ofAtg5orAtg7have limited fasting-induced steatosis.45,46
3.1.3.2.Lipolysis as an autophagy response to starvation.Lipid catabolism normally occurs via cytosolic lipases in response to fasting.Macrolipophagy also participates in intracellular hepatic LD degradation by upregulating LD delivery to lysosomes for subsequent lipolysis to generate free fatty acid (FFA) for mitochondrial oxidation into an energy source.19Inhibition of autophagy by 3-MA orAtg5knockdown in hepatocytes increases baseline TG content,while hepatic TG levels accumulate in liver-specificAtg7null mice after starvation.19It is stipulated that macroautophagy-mediated lipid biogenesis with sequestration of FFA released during starvation into the LDs to generate starvation steatosis is a time-restricted adaptive response to protect against hepatic lipotoxicity.43The packaged LDs subsequently undergo macroautophagy lipolysis to generate the needed energy substrates.
Hepatocyte LD catabolism in nutrient-poor cell culture can additionally occur independently of macrolipophagy.47Nutrient depletion activates hepatic CMA.48CMA-deficient hepatocytes derived from liver-specificLAMP2-null mice have advanced starvation steatosis with insensitivity to lysosomal capture,LD hydrolysis,and FA oxidation.20,49Despite intact macroautophagy,CMAdeficient liver exhibits more severe starvation steatosis,abnormal transition from lipid to carbohydrate catabolism,poor hepatic glycogen storage,and hypoglycemia,illustrating the importance of the overlapping and compensatory functions of macroautophagy and CMA in the nutrient regulation with starvation.19
The ER compartment regulates transmembrane and secreted proteins to coordinate cell metabolism.With ER stress events such as starvation,hypoxia,and oxidative injury,accumulated misfolded proteins can be degraded by activation of the two proteasomal UPR and the UPR-assisted ER-phagy systems.50Given the role of ER in lipid synthesis,UPR-assisted ER-phagy is implicated in ER nutrient response.ER-phagy activity increases during amino acid deprivation in mice liver,with paradoxical intrahepatic lipid accumulation.51Suppression of autophagy increases hepatic ER stress while restoration of autophagy improves ER stress while reducing TG contents.52
Mitophagy,a venue for the removal of damaged mitochondria,is also implicated in hepatocyte starvation response.Its activation in cultured hepatocytes under nutrient starvation medium paradoxically inducesde novolipogenesis from the released FFA.43It is also a protective response to sequester and degrade mitochondrial deoxyribonucleic acid(DNA),which is inhibited in the presence of 3-MA.53
3.1.3.3.Mechanisms of autophagy regulation.The mechanisms by which macroautophagy is involved in fat nutrient sensing by hepatocytes are multifold.Autophagy gene expression is regulated during the fasting state by transcription factor EB (TFEB),a master transcriptional regulator of starvation-induced autophagosomeand lysosomal-related genes;and during the fed state by the FA sensing nuclear hormone peroxisome proliferator-activated receptor alpha(PPARα),PPAR coactivator 1,and the bile acid sensing nuclear hormone farnesoid X receptor (FXR).54-56PPARα and FXR have competing binding sites on several autophagic gene promoters includingLC3with preferential PPARα binding during fasting to induce lipophagy,reduce TG levels and increase FA metabolism;while increased FXR binding in the fed stages suppresses autophagy.57-59In support of the link between autophagy and PPAR in FA metabolism,deficiency of Vps5 (a regulatory subunit of Class III PtdIns3K critical to phagophore formation)depletes PPAR transcriptional activity and impairs mitochondrial FAoxidation in primary hepatocytes,as well as in the liver ofVps5conditional null mice.60SIRT1 also directly regulates lipophagy by deacetylating the Atg proteins.SIRT1 overexpression in primary hepatocytes enhances autophagy lipophagic Palatin-like phospholipase domain-containing 2(PNPLA2)protein for LD catabolism and FA oxidation,an event which is blocked by inhibitor CQ.45As previously mentioned,Atg14is a direct target gene of the FoxO transcription factor,a transcriptional regulator of lipid metabolism.34In fasting mice,CRY1 is degraded by Atg7 in a timerestricted fashion to regulate lipid metabolism in primary hepatocytes and mouse liver.34
3.1.3.4.Summary.Therefore,the hepatic autophagy response follows the physiological circadian response of the liver to nutrient sensing.In close coordination with the clock proteins,hepatic autophagy increases gluconeogenesis to generate energy substrate in the fasting state.Upregulation of macroautophagy and CMA also induces LD hydrolysis by mechanisms such as transcriptional regulation by TFEB,FoxO1,SIRT1,and PPARα to increase FFA generation.The response is complex,time-restricted as seen with early starvation steatosis,and diverse as seen with ER stress-and mitophagy-associated paradoxical enhancement of lipogenesis under amino acid deprivation conditions.More studies are needed to understand the interplay between the various autophagy pathways in the combined responses to nutrients.
3.1.4.Autophagy,aberrant fat metabolism in non-alcoholic fatty liver disease and non-alcoholic steatohepatitis
3.1.4.1.Normal lipid metabolism.The liver is the dominant organ for the synthesis of TGs,steroids,and phospholipids to maintain cellular structural integrity and to regulate metabolic and signaling activities for digestion,energy generation,hormonal,and neurological function.
TGs (also referred to as triacylglycerols) come from either dietary sources or fromde novosynthesis.They are stored in hepatocytes as cytoplasmic LDs for subsequent TGs catabolism into FFAs either by cytosolic lipases or by macrolipophagy lysosomal lipases.
Cholesterol is the building block for steroids (sex steroids,corticosteroids,bile acids,etc.).Cholesterol is synthesized byde novosynthesis in the ER;where after detoxification by esterification,it is targeted to the plasma membrane or is packaged into LD in the ER for release as circulating lipoproteins;or is converted into bile acids in hepatocytes to meet the body digestive and detoxification needs.Cholesterol conversion into bile acid synthesis is the major route for cholesterol disposal.
Phospholipids are primarily synthesized in the ER of hepatocytes and are the major cellular membrane constituents for intercellular signaling.
Given the central role of the liver in fat metabolism,dysregulation of hepatic lipophagy is a major underlying factor for pathologies with underlying abnormal accumulation of intrahepatic fat.They most commonly include obesity-related non-alcoholic fatty liver disease (NAFLD) where excessive dietary fat consumption is a major contributor to intrahepatic lipid overaccumulation.In the setting of extrahepatic metabolic stress (such as diabetes)and intrahepatic stress (such as chronic endotoxemia),along with concurrent inflammation and immune dysfunction,a steatotic liver is the prerequisite for NAFLD progression to non-alcoholic steatohepatitis(NASH)with NASH risks of chronic fibrosis,cirrhosis,and hepatocellular carcinoma (HCC).61-63
3.1.4.2.Lipid overfeeding.When hepatocytes are overfed with TG in culture as a model of fatty liver disease,macrolipophagy is lipolytic,reducing intracellular lipid accumulation in response to autophagy inducers such as rapamycin.64Inactivation of lysosomal LD hydrolysis in hepatocytes impairs the clearance of TG and cholesterol,increases TG accumulation with lipid loading,and is reversed with autophagy inhibition by 3-MA treatment or inAtgknockdown.19,43,65In mice on a high-fat diet(HFD),enhancing autophagy through rapamycin treatment or byAtg7overexpression reduces steatosis.52,66
The intracellular toxicity of free cholesterol (FC) is neutralized by conversion to cholesterol esters stored in LD.When loaded with FC,lysosomal function is impaired with a dose-dependent increase of p62 in HepG2 cells or primary hepatocytes,leading to accrued intracellular FC contents.Stimulating bile acid synthesis by conversion from cholesterol reduces intracellular FC,stimulates mTOR autophagy,and reduces steatosis,while FC accumulation sensitizes the cells to hepatic injury of NASH-like pathology.67,68
Hepatocyte LD metabolism is also CMA-regulated.Dietary lipid or cholesterol overfeeding suppresses hepatic CMA.69CMAdeficient hepatocytes derived from liver-specificLAMP2-null mice display enhanced HFD-induced hepatosteatosis,reduced lipid catabolism,and hypoglycemia from impaired liver carbohydrate metabolism.20
ER-phagy is involved in hepatic lipid metabolism as previously mentioned.In rat liver,an HFD which induces fatty liver is also associated with increases in the expression of ER phagy receptorFAM134Bexpression along with upregulation of tumor necrosis factor (TNF) and ER stress index.70ER transmembrane proteins vacuole membrane protein 1 and transmembrane protein 41B(VMP1/TMEM41B) regulate lipid transport for phagophore elongation and are thus critical for autophagosome formation.71Mice with hepatic deletion ofVmp1have impaired hepatic phospholipid synthesis and lipoprotein secretion with associated mitochondrial dysfunction and susceptibility to NASH.72-74
Autophagy is also dysregulated in lipid storage diseases related to genetic disorders of phospholipid metabolism with adverse effects on the liver,such as defective sphingolipid storage in Gaucher disease and impaired sphingomyelin metabolism in Niemann-Pick disease.75-77
3.1.4.3.Lipopolysaccharide and fat metabolism.Lipopolysaccharide(LPS) is an important part of any discussion of abnormal fat metabolism since endotoxemia from the accumulation of this gutderived endotoxin impairs hepatic lipid metabolism.LPS suppresses FA oxidation,thus inducing acute and transient lipid accumulation in mouse liver.78Since the LPS hepatic effects are not just limited to autophagy but also manifest as endotoxemic sepsis and inflammation,LPS-associated hepatic autophagy responses are not just restricted to lipid metabolism (seesection 3.2.1 on autophagy and sepsis).In vitroandin vivo,a single high dose of LPS induces autophagy for 24 hours,and increases FA uptake with upregulation of PPAR and FA synthesis-associated genes along with accumulation of intracellular lipids.79Consistent with the link between LPS and lipophagy,the endotoxin level correlates with the extent of steatosis in the fatty liver.80It is noteworthy that the liver is normally exposed to low amounts of gut-derived LPS without pathological consequences.LPS-exposed hepatocytes are,however susceptible to cholesterol-induced ER stress,TNF and Fasdependent apoptosis and necrosis.68
3.1.4.4.Abnormal lipid metabolism and NASH.Intrahepatic lipid over-accumulation has adverse consequences.When exposed to FFA,lipid accumulation worsens in hepatocyte cell lines and in primary hepatocytes with associated impaired autophagy,increased ER stress,hepatic inflammation,and cell death.70,81-83In vivo,mice with liver-specificAtg7knockout not only develop hepatomegaly and increased TG accumulation with lipid supplementation,but also develop hepatic inflammation and apoptosis.19Pharmacologic orAtg5,Atg7 genetic inhibition of autophagy exacerbates FAassociated lipid accumulation and inflammation.These defects are improved with autophagy inducers such as rapamycin which alleviates ER stress and relieves steatosis and cell death.81-83
Altogether,the data implicate steatosis as a minimal requirement for NASH disease progression.While autophagy upregulation may alleviate the lipid burden,once a threshold of lipid accumulation is reached,lipotoxicity increases hepatocyte susceptibility to oxidative and ER stress and cell death.This provides the mechanistic basis for current observations of NASH evolution from NAFLD.Under conditions of chronic diet-induced gut-barrier permeability,with consequent increased endotoxemia,intrahepatic overexposure to LPS in a metabolically stressed fatty liver stimulates an aberrant innate immune response that recruits intrahepatic inflammatory monocytes and cytokines,leading the liver to transition from benign steatosis to inflammatory steatohepatitis.84,85In vivo,in support of the importance of autophagy in the abnormal liver response to lipid,steatotic liver of mice on HFD show evidence of impaired autophagy and NASH-like pathology.83Patients with NAFLD and NASH pathologies have impaired autophagy,intrahepatic lipid overload,ER stress,and hepatocellular apoptotic injury.83,86The extent of autophagy defect also correlates with the severity of steatosis in human NAFLD and in its progression to NASH.87Age-related autophagy defect in the liver is similarly associated with intrahepatic lipid accumulation.88
3.1.4.5.Summary.Thus,lipophagy is a major autophagy pathway in the liver reprogramming response to abnormal lipid metabolism that can generally be linked to survival by increasing lipid droplet breakdown to reduce the burden of lipid overload.Macroautophagy and CMA have distinct responses to excess lipid or LPS exposure.Their differential contribution needs further investigation.With excessive steatosis such as in NAFLD,impaired autophagy alters the normal hepatic lipophagy response,which,in a metabolically stressed fatty liver,can worsen the lipid burden to induce lipotoxic hepatocellular injury as in NASH.
3.1.5.Autophagy,bile acid and cholestasis
Cholestatic liver diseases and cholangiopathies (bile duct diseases) all share the features of impaired bile acid secretion by hepatocytes and BECs,necrotic and apoptotic hepatocellular death from the toxic supraphysiological level of bile acids,and secondary reactive inflammation and fibrogenesis which are precursors to cirrhosis and cholangiocarcinoma (CCA).89,90
Bile acids are essential for the intestinal digestion of dietary fat and fat-soluble nutrients,and for intrahepatic cholesterol,endobiotic and xenobiotic metabolism.In hepatocytes,bile acids are synthesized from cholesterol by the cholesterol 7a-hydroxylase(Cyp7a1)enzyme.Bile acids activate the nuclear hormone receptor FXR,which in turn activates downstream mediators to repressCyp7a1transcription as feedback inhibition of bile acid synthesis in the fed state or during cholestasis when bile acids are exceedingly abundant in the liver.The latter is an important mechanism for reducing intrahepatic bile acid content and attenuating hydrophobic bile salt injury.91Cyp7a1is also negatively regulated through the gut-liver cross talk.Intestinal FXR induction of intestinal fibroblast growth factor 15 (rodent Fgf15 or the human homolog FGF19 which binds FGF receptor 4 on hepatocytes) repressesCyp7a1via the mitogen-activated protein kinase (MAPK)pathway.92
Hydrophobic bile acids are cytotoxic.93They impair autophagy flux in primary hepatocytes and in hepatocyte cell lines at several steps of the autophagy flux,from autophagosome biogenesis,autophagosome-lysosomal fusion in a time-and dose-dependent manner,to lysosomal proteolysis.57,94,95This autophagy defect is associated with increased hepatocyte susceptibility to bile acidinduced cell deathin vitro,as well asin vivofollowing bile duct ligation (BDL).96Consistent with these findings,many adult patients with cholestatic conditions exhibit histological evidence of impaired hepatocellular autophagy.57,97In contradiction to these observations in adult cholestatic livers,pediatric biliary atresia liver samples show increased autophagy and expression of mitophagy genesATG5,ATG7,PTEN-induced putative kinase 1(PINK1),… .98Furthermore,in zebrafish with bile salt export pump (Bsep) deficiency and in patients withBSEPdeficiency that develop cholestatic hepatocyte injury,AtgsBeclin,Atgare upregulated.99Induction of autophagy with rapamycin rescues bile flow in theseBsepmutant zebrafish,suggesting thatin vivo,dysregulated autophagy may worsen some conditions of human cholestasis.
The mechanisms by which bile acids suppress autophagy are multifold.100Bile acids repress autophagy flux in an FXRdependent manner by stimulating FXR;and through FXR,by inducing Rubicon,a direct FXR target gene and a known inhibitor of autophagosome maturation.57Silencing of Rubicon in HepG2 reverses bile acid inhibition of autophagy.While the expression of Rubicon is elevated in the liver of cholestatic patients,it is decreased in non-cholestatic patients when treated with the anticholestatic agent ursodeoxycholic acid.57FXR also regulates the transcriptional activities of other ATGs (e.g.,LC3),in further supporting bile acid-mediated FXR regulation of autophagy in hepatocytes.58Consistent with this,FXR knockdown in HepG2 cells leads to increases in their resistance to bile acid inhibition of autophagy.57As previously mentioned,the bile acid-dependent FXR promoter binding site overlaps with that of nuclear hormone sensor PPARα which conversely activates autophagy,as a reminder of the liver nutrient status differential effects on hepatocyte autophagy.58To further complicate autophagy activities in hepatocytes,despite the loss of FXR expression,autophagy is blocked in cholestatic FXR null mice,suggesting that bile acid overload in cholestatic FXR null mice can suppress autophagy in an FXR-and Rubicon-independent manner.59,94Another described mechanism by which bile acids suppress autophagy in primary hepatocytes is through FXR-mediated Fgf5/FGF19 epigenetic repression of ATG,or by inhibition of transcription factor E3 (TFE3) which has a broad regulatory function on autophagosome formation and phagosomelysosome pathways.95,101
Regarding thein vivoeffect of autophagy on cholestasis,cholestasis is more advanced in mice with conditional deletion ofAtg5orAtg7in hepatocytes,while mice with tamoxifen-inducibleAtg7deletion develop time-dependent worsening of cholestatic injury with tamoxifen treatment.CQ inhibition of autophagy lysosomal activity worsensin vitrobile acid-induced hepatocyte apoptosis/necrosis and exacerbatesin vivobile acid-induced injury by BDL.102In support of the mentioned data linking impaired autophagy with worsening of bile acid-mediated hepatocyte injury and upregulation of autophagy as a protective response against bile acid injury,induction of autophagy by mTOR1 inhibitor Rapamycin or nerve growth factor enhances the viability of bile acid-exposed hepatocytes,while treatment of BDL mice with Rapamycin reverses bile acid-induced liver injury.97,102
With regard to the role of BEC in cholestasis,BEC exposed to serum starvation or hydrophobic bile acids develops mitochondrial and ER stress,senescence,and dysregulated autophagy.103BEC senescence is attenuated with p62 knockdown.104Consistent with thesein vitrodata,cholestatic liver samples from primary biliary cirrhosis show ultrastructural evidence of impaired autophagy in the senescent and damaged BECs.104
Bile acids,however,have the reverse survival effect on nonepithelial cells.HSC cell lines treated with bile acids in culture proliferate,become transactivated,secrete collagen,and increase autophagy activity in a PI3K dose-dependent manner.105
Therefore,the autophagyin vivohepatic responses during cholestasis are cell-specific and complex because of the nonautophagic toxic effects of free bile acids on epithelial cell function and the secondary hepatic response to cellular stress.It is generally impaired in the liver epithelial cells when exposed to hydrophobic bile acids from FXR-mediated inhibition of autophagy genes through Rubicon or Fgf5/FGF19,or from bile acidsuppression of TFE3 in hepatocytes;with suppression of autophagy being detrimental to bile acid-mediated liver injury during cholestasis.Hydrophobic toxic bile acids,however also cause inflammation,ER stress,mitochondrial ROS damage,and hepatocyte cell death.106,107Inhibition of ROS stress in bile acid-treated hepatocytes relieves bile acid cytotoxicity and reverses the defect in autophagy through increased expression and transcriptional activity of TFE3.108Furthermore,treatment with Rapamycin to enhance autophagy reduces the associated ROS and cellular injury.102Thus,in the chronic stages of cholestatic injury,thein vitrosuppressive effect of bile acids on hepatocyte autophagy may be counteracted by thein vivoROS-induced autophagy as secondary effects of bile acid overload,with protective effects,emphasizing the importance of considering the context in which autophagy acts in cholestatic liver.109
3.2. Autophagy and the liver response to inflammation
3.2.1.Autophagy and the liver response to sepsis
Sepsis is characterized by a systemic inflammatory response to bacterial infection,increased oxidative and ER stress,and mitochondrial dysfunction that leads to widespread cell death and multiorgan dysfunction.110The hepatocyte autophagy response is activated within 24 h of LPS endotoxin treatment.79In experimental sepsis models of intestinal injury and bacterial infection by cecal ligation and puncture,the endotoxemia response is characterized by upregulation of ROS,mitochondrial damage,and hepatocellular apoptosis,along with increased mitochondrial biogenesis.111-113Autophagy activation in LPS-treated hepatocytes and septic liver is PtdIns3K-dependent.112As previously mentioned,LPS induces lipid accumulation in the mouse liver.78,79Inhibition of LPS-induced autophagy with CQ aggravates intracellular lipid accumulation,increases hepatic inflammation in the mouse liver,and worsens survival.79
Similarly,mice with conditional loss ofAtg5orAtg7in the liver exhibit severe mitochondrial,ROS,and hepatocellular apoptotic damage with accelerated mortality,consistent with the protective role of autophagy in the removal of damaged mitochondria and in the restoration of mitochondria health through mitochondrial biogenesis to enhance hepatocyte survival.114-116The nature of the autophagy response to LPS is temporal,with reported activation within 3-6 h of cecal ligation and puncture,followed by a decline and loss by 18 h in the later stage of sepsis that is associated with worsening hepatocyte cell death and liver synthetic dysfunction.117
Inin vitroandin vivomodels of acute liver failure by LPS/Dgalactosamine (D-GalN) cotreatment,LPS may regulate hepatic autophagy through the inhibitory action of LPS-induced hepatocyte microRNA (miR)-19a on AMPK signaling.118Since LPS is a known ligand for pattern recognition by toll-like receptor (TLR) and TLR9 that trigger the inflammatory response underlying sepsis-induced mortality,TLR may participate in the regulation of hepatic autophagy.In support of this,sepsis-induced autophagy is suppressed inTlr4null mice.112,113Furthermore,Tlr9 knockdown in hepatocytes inhibits LPS-induced mitochondrial biogenesis which is also impaired inTlr9rmutant mice,worsening the hepatic injury.It is unclear how TLR4 and TLR9 regulate LPS-and sepsis-related autophagy,but mitochondrial DNA has been reported to bear sequence motifs that can be recognized by and can serve as ligands for TLR9.119Of note,older mice have reduced liver autophagy compared to young mice,thus displaying worse sepsis-related liver damage.120,121This may be related to diminished AMPK function or diminished telomerase activity in older mice.121,122
Macrophage autophagy in resident Kupffer cells or infiltrating monocytes contributes to the inflammatory response to LPS and infection and is also an important liver response to sepsis.LPS treatment of mice with fatty liver from an HFD or with D-GalN acute toxin-induced hepatitis impairs macrophage autophagy and promotes macrophage polarization toward the proinflammatory M1 phenotype,which worsens LPS-induced hepatocellular injury.123,124This is exacerbated in macrophage-specificAtg5null mice.LPS-induced macrophage mitochondrial dysfunction in mice withLC3IIandBeclin 1deficiency increases mortality.125
Liver sinusoidal endothelial cells (LSECs) regulate hepatic sinusoidal integrity,blood flow,and leukocyte-liver cell crosstalk.With liver injury,LSEC undergoes defenestration.126Autophagy is rapidly upregulated in LPS-treated,culture-starved LSEC and in mice with carbon tetrachloride (CCl4)-induced liver injury,with LSEC loss of cellular integrity and endothelial-mesenchymal cell phenotype transition,a phenomenon that is linked to hepatic fibrogenesis.127,128
Altogether,the available data suggest that autophagy is differentially regulated with LPS/sepsis in the liver epithelial and nonepithelial cells with diverse effects.Hepatocyte autophagy has a time-dependent response to LPS or sepsis-induced liver injury with a protective effect when it is upregulated,while impairment in the chronic phase is associated with hepatocyte cell death and poor outcome that has been linked to the miR-19a and TLR pathways.During sepsis,while autophagy downregulation in hepatic monocytes promotes their transition to the inflammatory phenotype,its upregulation in LSEC is linked to fibrogenesis,illustrating the complex cellular interactions in the liver during inflammation and the importance of context when investigating the hepatic autophagy response.
3.2.2.Autophagy and alcohol-associated liver disease
Alcohol-associated liver disease encompasses a spectrum of clinical presentations,from fatty liver(steatosis)to steatohepatitis,which is characterized by the induction of hepatic oxidative stress,mitochondrial injury,and inflammation from the toxic alcohol metabolites.
The effect of alcohol on liver autophagy depends on whether the exposure is acute or chronic.129,130A single high dose of alcohol induces upregulation of Atgs in primary hepatocytes.131Increased autophagy in alcohol-treated hepatocytes selectively removes the accumulated damaged mitophagic vesicles and lipid droplets.132This is suppressed with 3-MA inhibition of PtdIns3K,and with NAcetyl cysteine treatment,consistent with the role of ROS in alcohol-induced mitophagy.Mice acutely exposed to alcohol similarly exhibited increased autophagy flux,with increased lipophagy and microsteatosis,a known histologic association between alcoholic hepatitis and pathological steatosis.In these mice,pretreatment withAtg7small interfering RNA (siRNA) or the autophagy inhibitor CQ suppresses autophagosome formation and worsens hepatocellular apoptosis.129,132FoxO3a is a forkhead transcription factor that regulates cellular apoptosis,oxidant stress,cell renewal,and DNA repair.FoxO3 nuclear localization increases in primary hepatocytes and the liver with acute alcohol treatment,while FoxO3 knockdown suppresses alcohol-induced autophagy genes and exacerbates primary hepatocyte apoptosis.The same phenomenon was reported in FoxO3 null mice which exhibit hepatocellular injury.131The data suggest that autophagy induction is a protective mechanism to alleviate acute alcohol-induced hepatic apoptotic injury.
In contrast,autophagy and lysosome biogenesis are disrupted in mice with chronic alcohol exposure.Autophagy is impaired at the autophagosome-lysosome fusion stage in hepatocytes and HSCs,characterized by LAMP1,LAMP2,and transcription factor EB(TFEB) suppression in the experimental model of alcoholic injury by acute-on-chronic and chronic alcohol-feeding;as well as in liver samples from human alcoholic liver disease.130,133Increasing autophagy flux with LAMP2 improves alcohol liver injury,while lysosomal inhibition by CQ treatment worsens alcoholic liver injury.130,133With chronic alcohol feeding,impaired LAMP2-mediated autophagy flux and hepatocellular injury are characterized by hepatic FFA accumulation and increased ER stress that can be reversed with Rapamycin inhibition of mTOR signaling.133-135Mice defective in mitophagy by deletion of Parkin (which is essential to mitochondrial membrane recruitment of the cargo for engulfment by the autophagosome) also have worse alcoholinduced mitochondrial and oxidative stress damage,stress steatosis,and liver injury during alcohol binge and chronic feeding.136,137In old mice,defective autophagy receptor p62 function impairs hepatic lipid processing with alcohol feeding,leading to obesity and liver inflammation.138
There are multiple pathways for the autophagy defects leading to alcohol-induced steatosis,including impairment of mTOR;of TFEB regulation of lysosomal biogenesis;of lysosome-associated membrane protein LAMP1 and LAMP2 or small GTPase Rab7 regulation of autophagosome-lysosome fusion or dynamin 2 activity.133,134,139,140
miRs are non-coding small endogenous RNAs that regulate posttranscriptional gene expression and are increasingly recognized for their regulatory function in inflammation and cell survival.Alcohol-induced hepatocyte exosome miR-155 has been linked to the suppression of autophagosome-lysosome function in hepatocytes,withmiR-155-deficient mice showing restored LAMP1 and LAMP2 and being protected from alcohol-induced exosome damage.141Consistent with the above findings,liver samples from patients with alcoholic liver disease show increased miR-155 in exosome fractions,as well as evidence of impaired autophagy and lysosomal function with p62 accumulation and reduced LAMP1,LAMP2,and TFEB.142
Thus,autophagy upregulation in hepatocytes is protective against acute alcohol-induced hepatitis,while dysregulation in the chronic setting can be linked to progressive injury and chronic alcoholic hepatitis.
Alcohol also affects HSC function by triggering autophagy and stimulating oxidative stress and transactivation in culture into myofibroblast (MFB),suggesting that autophagy in HSCs is a contributor to alcohol-induced liver fibrosis.HSC autophagy inhibition conversely can reverse alcoholic fatty liver in the mouse model of alcoholic liver fibrosis.143
In conclusion,these data show that autophagy is a time-related survival response to alcohol-associated liver injury in hepatocytes.In the acute phase,autophagy dampens hepatocellular damage.In the chronic phase,it is impaired and adversely affects hepatocyte survival,while activation of HSC induces hepatic fibrosis.Unrelieved inflammation and sustained HSC activation are major fibrogenic factors in chronic alcoholic-associated liver disease,cirrhosis,and progression to HCC,thus providing the link between chronic alcoholic hepatitis and HCC.144
3.2.3.Autophagy and viral hepatitis
The discussion of autophagy will be restricted to viral hepatitis caused by hepatitis B virus(HBV)and hepatitis C virus(HCV)since they are the most common pathogens implicated in chronic viral hepatitis,and account for the highest worldwide cause of cirrhosis and HCC.
3.2.3.1.HBV and autophagy.The HBV particles consist of envelope surface antigens known as hepatitis B surface antigen(HBsAg);the hepatitis B core antigen (HBcAg) that forms the nuclear capsid containing the viral genome;the HBp DNA-dependent DNA polymerase;and the non-structural protein-hepatitis B virus X protein(HBx).HBx is the major underlying player in HBV-related pathologies through its effects on viral replication and chronic infection,on disruption of the immune response,and on induction of the ROS response to perpetuate the antiviral chronic inflammatory state in the liver as a risk factor for hepatic fibrosis,cirrhosis,and HCC.145
The preponderance of data derived from culture systems suggests that while HBV stimulates the normal hepatocyte xenophagic flux against pathogens,it can hijack the autophagy process to evade the antiviral response and survive in infected hepatocytes.The mechanisms include HBs activation of ER stress-induced Beclin1 and ATG5 autophagy in hepatoma cell lines to enhance their replication,or HBs interaction with Rab7 small GTPase to block autophagosome-lysosome fusion.146,147Alternatively,HBx protein activates PI3KC3 activity;increasesBeclin-1transcriptional activation through c-Myc-dependent X-linked inhibitor of apoptosis protein (XIAP,X chromosome-associated apoptosis protein)-nuclear factor-kappa B (NF-kappa B) pathway;increases PI3KC3/Beclin-1 interactions to promote viral DNA replication;and impairs lysosomal function to interfere with viral elimination.148-151The HBV core particle also associates with the Atg5-12/16L1 complex in hepatocyte cell lines to subvert phagophore formation,or binds to the phagophore and autophagosome membranes to promote HBV assembly and trafficking.147,152,153Consistent with thesein vitrofindings,HBV transgenic mice with hepatocyte-specific deletion ofAtg5have undetectable HBV viral DNA in the livers,and ATG16 expression levels are higher in tumor cells compared to non-tumor cells from infected HBV liver.154,155
3.2.3.2.HCV and autophagy.HCV carries the structural envelope glycoproteins E1 and E2 for cell entry and immune cell interactions;the core protein;and several non-structural(NS)proteins including p7 ion channel,NS2,NS3,NS4A,NS4B,NS5A,and NS5B,all of which are important to viral replication and viral particle assembly.Many autophagy steps are sequentially affected during early HCV infection,as individually silencing Beclin-1,ATG4B,ATG12 or ATG5 interferes with viral replication.HCV can,however,survive by multiple mechanisms,with HCV ER stress inhibition of mTOR1,promoting defective autophagosome-lysosome fusion;by HCV impairment of the nuclear erythroid 2-related factor 2/antioxidant response element (Nrf2/ARE) protective genes to promote a persistent ROS stress response;or p62-dependent impairment of HCV autophagic degradation.156-160HCV structural proteins can also enhance viral replication or viral production through the interaction of their NS3 unit with small GTPase,NS5B unit interaction with Atg5,and NS4B suppression of autophagosomelysosome through Rubicon.161-163The latter 2 events are,however,time-sensitive as shown by delayed restoration of the autophagy defect from the inactivation of the Rubicon by the UV radiation resistance-associated gene (UVRAG).HCV autophagy also hijacks the autophagosomes to dock its ribonucleic acid (RNA) replication complex to enhance its maturation and release;to subvert the antiviral innate immune response as silencing HCV-induced UPR complex or Atg5 suppresses interferon (IFN) and viral particle production in culture;or to inhibit mTOR1 signaling by IFNα-2a to increase the levels of secreted and intracellular HBs in primary hepatocytes and HCC cell lines.164-167
HCV NS5A protein has been reported to activate mitophagy by inducing mitochondrial degradation by parkin translocation to the mitochondria to enhance the survival of the infected cell,while the HCV core protein has the opposite effect on mitophagy.168,169
Beyond mediating the autophagy response to ROS oxidative damage from viral infection,chronic HBV or HCV infection also affects hepatocyte lipid and bile acid metabolism.170HCV selectively induces lipophagy in the infected cells to induce steatosis,a known histological finding in chronic hepatitis,with lipophagy being linked to supporting viral cell entry and replication.171,172Consistent with this,HCV liver samples show a correlation between the extent of autophagy and microvesicular steatosis,while autophagy suppression in HCV-infected cells was related to high intracellular cholesterol accumulation.173HBV also utilizes bile acid nutrient flux for survival,as reflected by its dependence on the bile acid transporter Na+/taurocholate cotransporting polypeptide(NTCP) for cellular entry,and on FXR for viral replication.174,175
3.2.3.3.Summary.The nature of the autophagy response to viral hepatitis is therefore complex and specific to the infecting organism and the life cycle of the virus.The initial induction of autophagy activation is part of the antiviral response for viral elimination and the antiviral inflammatory and immune response.However,viruses can manipulate autophagy in both directions to enhance viral formation,maturation,and trafficking,or to subvert viral elimination by either directly targeting the different steps of the autophagy machinery during the various phases of their life cycle,or by indirectly modulating autophagy through the liver antiviral oxidative and ER stress response.
3.2.4.Autophagy and chronic hepatic inflammation andfibrosis
Chronic inflammation and ROS stress are shared features of the liver fibrogenic response to hepatitis regardless of the underlying etiologies (toxins,virus,alcohol,fatty liver,autoimmune diseases,etc).176
Hepatic macrophages (resident Kupffer cells or infiltrating monocytes)are implicated in the fibrogenic response because they recruit inflammatory profibrotic mediators,and activate HSC myofibroblastic transformation.In mice with macrophage-specific deletion ofAtg5,the monocytes acquire increasedin vitroinflammatory and fibrogenic response to LPS,andin vivohepatic inflammation and cellular death during GalN/LPS or CCl4toxin liver injury.123,177,178These effects are reversed with interleukin-1 (IL-1)receptor inhibition,implicating the importance of the contribution of hepatic macrophage inflammatory properties in hepatic fibrogenesis,and the potential interplay of autophagy in the suppression of hepatic macrophage inflammatory response during hepatic fibrosis.178
HSC is,however,the most important cellular mediator of extracellular matrix (ECM) secretion and deposition that lead to scar tissue formation and hepatic fibrosis upon HSC activation and transdifferentiation into hepatic MFB,with the loss of LD content being correlated with the transactivation status of HSC while accumulation of LD being associated with HSC reversion of activation status.179Autophagy has a profibrotic effect on HSC as it activates HSC proliferation and transdifferentiation into MFBs.In freshly isolated HSC or cell lines,activation of cultured primary HSC by hypoxia,LPS,arsenic trioxide (As2O3),transforming growth factor-β1 (TGF-β1),or oxidant stress by hydrogen peroxide (H2O2)induces autophagy with HSC loss of cytoplasmic LD via ROS-Rab25-dependent pathway and HSC sensitization to TGF profibrotic effect.180-185Conversely,autophagy inhibitors such as bafilomycin A1 or LC3 orAtg7siRNA inhibit HSC-T6 cell line proliferation.Inhibition of autophagy with 3-MA or CQ andAtg5orAtg7knockdown interferes with LD turnover along with inhibition of HSC fibrogenic gene expression.180,185-188Accumulation of p62 with autophagy can also suppress p62-mediated activation of vitamin D receptor/retinoid X receptor α (RXRα) in HSC,suggesting the presence of autophagy auto-regulation in HSC fibrogenesis.189
The mechanisms for autophagy induction in HSC are numerous,including AKT (or protein kinase B)-mTOR and AMPK-ULK1 pathways in endotoxin-mediated bacterial translocation;calciumdependent activation of AMPK/mTOR and protein kinase C-theta(PKCθ)pathways;plasmacytoma variant translocation 1(PVT1)and miR-152 induced ATG14 during hypoxia;G-couple protein mediated c-Jun N-terminal kinase(JNK)and ATG12-5 conjugation;highmobility group box-1 (HMGB1) induction via the extracellular signal-regulated kinase (ERK)/JNK-MAPK and mTOR/signal transducer and activator of transcription 3 (STAT3) signaling pathways;PM2.5(a Fine particulate matter)regulation through PINK1/parkin signaling;RNA-binding protein ZFP36/tristetraprolin (TTP) destabilization of ATG16L1 to impair HSC cell death;TGF-β1 mediated small mother against decapentaplegic (SMAD) signaling through ERK and JNK;C/EBPa as HSC mitophagy regulator;and insulin-like growth factor binding protein-related protein 1 (IGFB-rP1) activation of the PI3K/Akt/mTOR signaling.180,181,183,190-195In vivo,increased autophagy activity was detected in HSC from mouse and human fibrotic livers,and inhibition of autophagy prevented HSC activation and liver failure.188
Hepatocytes or BECs undergo transformation during the ductular reaction (DR),where bipotential liver stem cells or hepatic progenitor cells (HPCs,also known as ductular or oval cells)differentiate into hepatocytesvs.reactive ductular BEC-like cells.196,197They also adopt epithelial-to-mesenchymal transition (EMT) with loss of epithelial cell polarity and acquisition of the mesenchymal myofibroblastic features that favor the fibrogenic response.128,198Both phenomena have been linked to fibrogenesis in the settings of chronic inflammation and sustained hepatocellular cell loss.Autophagy,however,differentially affects hepatocytes and BECs.In rat liver cirrhosis induced by 2-acetylaminofluorene and CCl4injury,or in the human cirrhotic liver,autophagy is activated in reactive ductular cells and induces EMT.199Autophagy suppression by bafilomycin A1 or siRNAAtg7knockdown reduces the DR and attenuates BEC differentiation into the mesenchymal cell phenotype,with alleviation of fibrosis.199In contrast,autophagy inhibits hepatocyte profibrogenic TGF-β induced EMT response,and mice with liver-specificAtg7depletion show increased fibrogenic liver with upregulation of α-smooth muscle actin (α-SMA) hepatic expression,consistent with autophagy role in maintaining hepatocyte in a differentiated state.200
As previously mentioned,LSEC is important to liver endothelial cell integrity.Autophagy is rapidly upregulated in LPS-treated,culture-starved LSEC and in mice with CCl4-induced liver injury.127LSEC-specificAtg5andAtg7null mice have aggravated hepatic inflammation and fibrosis with high-fat feeding or CCl4liver injury.201,202
Thus,autophagy upregulation in HSC in a chronic inflammatory microenvironment is the major factor for fibrogenesis with mixed participation from the liver epithelial cells in this response.
3.3. Autophagy and liver regeneration
3.3.1.Autophagy and hepatocellular proliferation
Partial hepatectomy (PHx) through 70% liver resection triggers hepatocyte proliferation and is the classical mouse model for liver regeneration.Autophagy is activated in the early course of PHx.Pharmacologic induction of autophagy accelerates hepatocyte cell cycle progression and proliferation,increases liver mass independently of proliferative cytokines IL-6 and TNF-α,and improves survival during near complete hepatectomy by 90%liver resection.Consistent with this,Atg7siRNA treatment suppresses liver growth and metabolic function during PHx.203,204Similarly,mice with liver-specificAtg5deletion have impaired mitochondrial function,reduced hepatocyte DNA synthesis with early senescence,and higher mortality with 90%hepatectomy.205Mice with CCl4-induced advanced liver fibrosis or NAFL also have impaired autophagy and liver regeneration during 50% hepatectomy.In these mice,treatment with the autophagy inducer verapamil partly restores hepatocyte proliferation and liver regeneration.206,207
The liver also undergoes different forms of organ regeneration as previously mentioned,especially in response to severe chronic injury.In response to chronic hepatic injury and major loss of functional liver mass,the liver undergoes DR where bipotential HPC liver stem cells differentiate into hepatocytes or reactive ductular BEC-like cells to counter massive epithelial cell loss.196,197Hepatocytes and BECs also undergo EMT during the liver repair process to achieve myofibroblastic fibrogenic transformation.128
In vitro,autophagy induction inhibits TGF-induced EMT in hepatocyte cell lines.Similarly,mice with liver-specificAtg7depletion have increased expression of hepatic mesenchymal indicators such as α-SMA,a marker of hepatic fibrosis activity.200All-trans retinoic acid-mediated autophagy is activated in rat HPC cell lines during the early stages of their differentiation into the hepatocyte cell lineage,while overexpression of Delta-like 1 homolog (DLK1) cell differentiation regulator in HPC increases autophagy and favors hepatocyte maturation.208,209Overexpression of the autophagy gene restores stem cell function,while autophagy inhibition impairs cell self-renewal,cell proliferation,and hepatic differentiation in culture,along with suppression of HPCs differentiation into hepatocytes in human and mice fibrosis injury models.210,211Loss of autophagy in hepatocytes suppresses HPC differentiation into the hepatocyte lineage to favor biliary-like HPC DR formation.212In vivo,mice with conditional deletion ofAtg5orAtg7in hepatocytes exhibit more prominent ductular reaction histology.102
Atg5 is highly expressed in biliary Cytokeratin(Ck)19+HPC,andin vitroinduction of autophagy by Rapamycin increases cultured oval biliary cell proliferation,while inhibition by CQ decreases cellular proliferation and increases apoptosis.213Autophagy is also increasedin vitroandin vivoin damaged BEC.In a rat model of liver fibrosis,autophagy suppression with bafilomycin A1 or siRNAAtg7knockdown reduces ductular cell acquisition of the mesenchymal cell phenotype.199
As previously mentioned,autophagy induction is closely linked to HSC proliferation and transactivation.105,143
Therefore,autophagy maintains hepatocytes in a proliferative differentiated state against EMT,while promoting BEC ductular transformation and stimulating HSC proliferation and dedifferentiation.However,the mechanisms by which autophagy differentially modulates HPC cell fate determination toward hepatocyticvs.BEC cell lineage are undetermined.
3.3.2.Autophagy,abnormal hepatocellular proliferation,and carcinogenesis
As previously mentioned,autophagy is upregulated with acute inflammation,while impaired autophagy with chronic oxidative stress worsens hepatic injury.The pathogenesis of HCC or CCA from BECs is still undelineated,but common associated factors in viral,autoimmune,alcohol-associated,non-alcoholic steatohepatitis in HCC progression are chronic cellular proliferation,chronic liver inflammation,chronic ER and oxidative stress,and activation of oncogenic signaling and mutations.63,214-217Similarly,CCA predisposing factors are chronic biliary infection and chronic bile duct autoimmune inflammation such as in sclerosing cholangitis or primary biliary cirrhosis.218Manipulation of the autophagy response has been used in many clinical trials for advanced unresectable HCC.Systemic first-or second-line drug combination treatment with molecule-targeted therapy of the tumor biology using multikinase inhibitors or immunotherapies have achieved limited outcomes because cancer chemoresistance and disease progression affect overall survival;and because of the heterogeneous underlying etiologies and tumor microenvironment.219Hepatic carcinogenesis has been linked to hepatocyte EMT where,in the setting of chronic inflammation and stress,hepatocytes acquire cancer stem-like cell characteristics and invasive properties to promote tumor formation and metastasis.128,220,221DR arising from HPC is also implicated as a risk factor for tumor development,with DR being linked to early onset HCC by one year of age,and to poor survival in human HCC because of the increased susceptibility of HPC to transformation and invasion.196,197In the chronic inflammatory and nutrient-poor microenvironment that promotes carcinogenesis in HCC and CCA,autophagy is likely to be dysregulated.
3.3.2.1.Autophagy function and its effects on HCC carcinogenesis or progression are inconsistent
3.3.2.1.1.Autophagy suppression in tumor development or progression.Diminished autophagy response underlies risks for HCC development in some settings.Mice withBeclin1mutations or hepatocyte-specific loss ofAtg5andAtg7spontaneously develop HCC.180,181Consistent with these findings,in human cirrhotic HCC liver,autophagy is demonstrated in the normal liver tissue adjacent to HCC foci,but is suppressed in HCC nodules.222Conversely,autophagy upregulation inhibits HCC progression.Quercetin induction of autophagy flux in HCC cell lines reduces their viability,induces apoptosis,and inhibitsin vivotumor growth.179Similarly,autophagy activation in CCA cells is AKT-mTOR dependent,inhibits cell proliferation,induces cell apoptosis,and sensitizes cells to chemotherapeutic agents.223
3.3.2.1.2.Autophagy inhibition in tumor suppression.Inhibition of autophagy with CQ,however,induces apoptosis in HCC cell lines.224Inhibition of autophagy with anti-tumor drugs or with CQ or 3-MA similarly reduces the viability of HCC cell lines,decreases TGF-β-induced EMT and invasiveness in cell lines,and improves patient survival.225-231Similarly,ULK1 knockdown inhibits HCC cell proliferation and tumor growth in the xenograft mouse model of HCC.232
In contrast,upregulation of CMA LAMP-2A in serum-starved HCC cell lines or in HCC xenografts enhances cell survival,whereasLAMP-2Aknockdown or CQ inhibition decreases cell viability.222-234Consistent with these findings,in human cirrhotic HCC livers,while macroautophagy is demonstrated in normal liver tissue adjacent to HCC foci but is suppressed in HCC nodules,LAMP-2 is uniformly upregulated in both tissue types,with an expression pattern concentrated to biliary canaliculi,implicating CMA as a tumor promoter.Thus,CMA activation in settings of impaired macroautophagy in the cirrhotic liver may favor tumor progression.226,235
3.3.2.1.3.The link between p62 and HCC is also complex.
Elevation in the expression of p62 as a marker of autophagy defect is seen in HCC cell lines but not in primary hepatocytes,with a dose-dependent clearance of p62 upon mTOR inhibition.p62 is also upregulated in foci of HCC in human cancer with a correlation with tumor aggressiveness.226p62 accumulation with its antioxidative Nrf2 cargo can drive tumorigenesis in HCC cell lines,human livers,and animal models of HCC,with p62 upregulation being linked to activation of Nrf2,mTOR1,and oncogenic c-Myc,which may protect the precancerous hepatocytes from cell death.236,237In contrast,deletion of p62 in mice with conditional loss ofAtg5orAtg7in the liver blunts the formation of HCC.Beyond its effects on hepatocytes,p62 also acts as an HCC suppressor with HSC-specific p62 ablation promoting myofibroblastic transformation with inflammation,fibrosis,and HCC progression.189
FoxO is a transcription factor that normally stimulates autophagy.Regarding CCA,knockdown ofFoxOin CCA cells decreases CCA cell survival.238,239
Hepatocyte-excreted exosomes can either suppress or activate autophagy to act as “onco-miRNA” to promote HCC progression,and to induce delayed HCC resistance to chemotherapeutic agents.240-242
Consistent with autophagy dichotomous effects on HCC,the expression of autophagy markers in HCC liver tissue is variable.In human liver HCC biopsy samples,autophagy is suppressed with p62 accumulation in tissue foci of HCC,but not in normal parenchyma;and high expression of p62 correlates with poor survival.212,224,243,244The loss of Beclin1 expression,along with the induction of hypoxia-inducing factor 1α (HIF-1α) expression,is associated with human HCC progression.245Similarly,tumors with low LC3 expression have a poorer prognosis.246However,other reports have demonstrated that tumors with high levels of LC3 expression are more aggressive and have a worse outcome.247,248
It is thought that the autophagy activation in the early stage of cancer may be protective in its effects on enhancing the antioxidant anti-inflammatory responses and the disposal of damaged proteins to suppress hepatocarcinogenesis.249,250However,in the microenvironment of well-established HCC where hypoxia,ROS,and ER stress response are accentuated,or during the tumor cell response to chemotherapy,autophagy is conducive to HCC or cancer stem cell proliferation and survival,and to increased tumor resistance to chemotherapy.251
3.3.2.2.Summary.Thus,the cell types,stage and chronicity of the disease,the underlying risks and genetic factors,and the microenvironment,highly influence autophagy intrahepatic biology,illustrating the complexity of the autophagy response in HCC and CCA.252-254
3.3.3.Autophagy in the aged liver
Hepatic autophagy activity decreases with aging.Consistent with autophagy effects on hepatocyte survival,hepatocyte mitosis after PH is suppressed in mice with liver-specific deletion ofAtg5,along with accumulation of ubiquitinated protein.255Suppression of autophagy reduces adenosine triphosphate (ATP) generation,impairs liver regeneration,and promotes liver senescence.205Aged hepatocytes exhibit poor generation and lysosomal clearance of autophagosome because of defective motor proteins dynein and kinesin family member C3.256In senescent hepatocyte cell lines,the autophagy response to oxidative stress is reduced with a switch of the cellular fuel source from B-oxidation to glycolysis,increased susceptibility to FA toxicity and reduced energy production.257In vivo,hepatic lysosome number,activity and substrates are reduced;AMPK function or irisin-dependent autophagy activity and telomerase activity are diminished in older mice,thus accounting for reduced liver autophagic flexus and worse sepsis-or ischemia reperfusion injury-and alcoholrelated liver damage.120-122,138,258
In summary,diminished liver autophagy function with aging increases the burden and clearance of misfolded proteins,impairs energy production and reduces hepatocyte proliferation and survival during the liver adaptive response to injury.
4.Targeting autophagy in human liver diseases
We are limiting the discussion of clinical trials to those registered on ClinicalTrials.gov that target autophagy in human liver diseases.There are 8 applicable studies (See Table 1).

Table 1 Description of registered clinical studies with autophagy-based intervention for liver disease.
Two observational studies characterized the intrahepatic autophagy response in clinical liver ischemia/reperfusion injury and NAFLD disease,and one interventional trial evaluated the effect of autophagy inhibition on viral clearance.
NCT02102971-Completed study in 2019.No available updated report.
This is an observational study to test if SIRT1,an autophagy inhibitor,is protective of liver ischemia-reperfusion injury in 41 subjects undergoing liver resection or transplantation for HCC.
NCT01988441-Ongoing study.
This is a prospective observational study to describe Atgs expression and compare Atgs genetic polymorphisms in NAFLD development for a 1 year follow-up in 800 obese subjects aged 6-18 years.
NCT02058173-Completed study in 2015.No available data.
This interventional pilot randomized controlled trial (RCT) targets 20 subjects aged 18-60 years old who had poor response to standard antiviral HCV treatment.They will be randomized to a 12-week course of autophagy inhibitor CQ or placebo to assess the primary outcome of viral clearance.
The following five studies are all interventional trials with one targeting CCA or four targeting advanced HCC (including four trials on autophagy inhibition with hydroxychloroquine (HCQ)-based studies and one study on autophagy inhibition with ezurpimtrostat-based study).As background,advanced or unresectable HCC respond poorly to current treatment approaches using vascular endothelial growth factor (VEGF) inhibitors,multikinase inhibitors (e.g.,sorafenib),and more recently,combination therapy with atezolizumab (tumor cell immune checkpoint inhibitor)-bevacizumab (VEGF inhibitor)(AB);with logoregional therapy as a supplement to systemic therapy.Transarterial chemoembolism (TACE) is a type of locoregional therapy where the arterial supply to the tumor mass is infused with chemotherapeutic agents and subsequently embolized with lipidiol to disrupt the tumor vascularity.AB is currently a first-line therapy for patients with unresectable HCC who are not candidates for locoregional therapy.259In spite of these interventions,cancer recurrence or ineffective response is common,in part because of the cancer cells’ ability to upregulate autophagy as a survival response to the nutrient-poor and hypoxic stress environment.251The trials described below address the study questions if the addition of autophagy inhibitors can enhance the tumor response to currently available treatments.
Autophagy inhibition with HCQ-based studies.
NCT04566133-Ongoing study (2022-Present).
This open-label,non-randomized phase 2 study targets 30 patients ≥18 years old with unresectable CCA tumor subtypes bearing the Kirsten rat sarcoma viral oncogene homolog(KRAS)mutations.They are to receive a combination of oral HCQ and trametinib (inhibitor of mitogen-activated extracellular signal-regulated kinase).Drug safety and 5-month tumor progression-free survival will be assessed.
NCT04873895-Ongoing study (2022-Present).
This is an open-label phase 1b safety determination study in 25 subjects ≥18 years old with liver-dominant metastases from colorectal cancer who have failed systemic chemotherapy.They are to sequentially receive(a)a 2-week combined course of oral Axitinib(a VEGF receptor kinase inhibitor) and autophagy inhibitor HCQ,followed by(b)monthly TACE treatment of the diseased liver lobe or segment until the entire liver tumor burden is treated,and (c)axitinib/HCQ treatment until toxicity develops.Subjects will be followed for 1 month and every 3 months for 12 months for toxicity,liver disease progression,progression-free survival,and overall survival.
NCT05842174-Pending.
This is a phase 1/2 RCT interventional study that randomizes 93 subjects ≥18 years old with intermediate-stage HCC to the sequential treatment of (a) TACE delivery of HCQ+lipidiol embolization followed by a 6 weeks course of oral HCQ,against (b) a control arm receiving TACE+lipidiol only and a 6 weeks course of oral placebo.Safety assessment and 6 months tumor progressionfree survival are the primary study goals.
NCT03037437-Ongoing study (2017-Present).
This open-label,non-randomized study enrolled 68 subjects≥18 years old with unresectable or metastatic HCC to receive HCQ and tyrosine kinase inhibitor sorafenib combination treatment.Dose safety and time to tumor progression at 1 year will be assessed.
Autophagy inhibition with Ezurpimtrostat-based study.
NCT05448677-Ongoing study (2022-Present).
This open-label,multicenter phase 1 trial will first assess the safety and dose determination of autophagy inhibitor Ezurpimtrostat and AB combination treatment in up to 12 subjects ≥18 years old with unresectable HCC;followed by a phase 2 efficacy phase that randomizes 184 subjects to AB+Ezurpimtrostat or AB alone for a 36 months progression-free survival as the primary outcome.
5.Conclusions
Our understanding of hepatic autophagy is rapidly changing and evolving.Future investigations addressing current knowledge gaps and conflicting data are needed.Published studies have been mostly limited to investigating a specific type of autophagy response in isolation,while autophagy activities in biological systems are overlapping and can occur simultaneously or in a compensatory manner.260Although the autophagy response at the cell-specific level under defined culture conditions is welldescribed,the in vivoeffects at the multicellular organ level can be multidirectional(Fig.3).For instance,induction of autophagy in hepatocytes increases hepatocyte proliferation to enhance the hepatic regenerative response to parenchymal injury and to hepatocyte loss with beneficial outcomes,yet promotes cancer stem cell survival and resistance to chemotherapeutic agents;while the same hepatic autophagy signals may stimulate HSC transdifferentiation into MFB to induce or worsen hepatic fibrosis.Broad differences between patients in their disease type,stage,and course with diverse underlying individual biological characteristics and microenvironment varying from simple inflammation to coexisting ER stress,fibrosis,....further complicate efforts in autophagy-based clinical interventions.
Autophagy is the driver of the liver cell function and survival in normal physiology and in adaptation to a broad spectrum of underlying liver pathologies,from fatty liver to hepatic inflammation,fibrosis,cell regeneration in cholestasis,sepsis,nonalcoholic/alcoholic-associated/viral hepatitis,and hepatocellular/cholangiocellular cancer.Autophagy is a ubiquitous phenomenon to both the liver epithelial and non-epithelial cells in normal and pathological conditions.While induction or upregulation of autophagy is generally a protective response to enhance cell survival,it also has diverse effects depending on the inducing signals and hepatic microenvironment.Continued investigations into cellspecific contributions to the autophagy response with considerations of disease context as the basis for thoughtful clinical trials are needed.
Authors contributions
AiXuan Holterman:Conceptualization;Supervision;Validation;Manuscript original draft preparation,editing and review.Trinh Van Le: Conceptualization;Data curation;Formal analysis;Graph design;Manuscript original draft preparation,editing and review.Nhung Hai Truong: Supervision and Writing -manuscript editing and review.All authors have read and agreed to the published version of the manuscript.
Declaration of competing interest
The authors declare that there is no conflicts of interest.
Acknowledgements
This work was supported by Prometheus USA.

- Liver Research的其它文章
- Dietary pattern and hepatic lipid metabolism☆
- In-depth analysis of de novo lipogenesis in non-alcoholic fatty liver disease: Mechanism and pharmacological interventions☆
- The contributions of bacteria metabolites to the development of hepatic encephalopathy☆
- Hepatocellular carcinoma recurrence: Predictors and management☆
- Unveiling the effect of estrogen receptors in alcoholic liver disease:A novel outlook☆
- Sequential ultrasound molecular imaging for noninvasive identification and assessment of non-alcoholic steatohepatitis in mouse models☆