
Environment drives the co-occurrence of bacteria and microeukaryotes in a typical subtropical bay*
2023-12-23 10:16YifanMALingfengHUANGWenjingZHANG
Yifan MA, Lingfeng HUANG, Wenjing ZHANG,**
1 State Key Laboratory of Marine Environmental Science, Marine Biodiversity and Global Change Research Center, College of Ocean and Earth Sciences, Xiamen University, Xiamen 361102, China
2 Xiamen Key Laboratory of Urban Sea Ecological Conservation and Restoration, Xiamen University, Xiamen 361102, China
3 Fujian Key Laboratory of Coastal Pollution Prevention and Control, Xiamen University, Xiamen 361102, China
4 Key Laboratory of the Ministry of Education for Coastal and Wetland Ecosystems, College of the Environment and Ecology,Xiamen University, Xiamen 361102, China
Abstract The co-occurrence of bacteria and microeukaryote species is a ubiquitous ecological phenomenon, but there is limited cross-domain research in aquatic environments.We conducted a network statistical analysis and visualization of microbial cross-domain co-occurrence patterns based on DNA sampling of a typical subtropical bay during four seasons, using high-throughput sequencing of both 18S rRNA and 16S rRNA genes.First, we found obvious relationships between network stability and network complexity indices.For example, increased cooperation and modularity were found to weaken the stability of cross-domain networks.Secondly, we found that bacterial operational taxonomic units (OTUs)were the most important contributors to network complexity and stability as they occupied more nodes,constituted more keystone OTUs, built more connections, more importantly, ignoring bacteria led to greater variation in network robustness.Gammaproteobacteria, Alphaproteobacteria, Bacteroidetes, and Actinobacteria were the most ecologically important groups.Finally, we found that the environmental drivers most associated with cross-domain networks varied across seasons (in detail, the network in January was primarily constrained by temperature and salinity, the network in April was primarily constrained by depth and temperature, the network in July was mainly affected by depth, temperature, and salinity, depth was the most important factor affecting the network in October) and that environmental influence was stronger on bacteria than on microeukaryotes.
Keyword: co-occurrence network; cross-domain; network stability; network complexity; subtropical bay
1 INTRODUCTION
In natural ecosystems, organisms are interconnected via predation, competition, mutualism, parasitism,and other interactions to form complex networks(Faust and Raes, 2012).Co-occurrence networks generated from microbial DNA datasets using mathematical and statistical methods (e.g., correlationbased methods) (Faust and Raes, 2012) can be used to infer the interactions among co-occurring members and their relationships with environmental factors.Nodes and edges in networks usually represent organisms and statistically significant associations between organisms, respectively.Therefore, networks can reflect species interaction and dynamics of ecosystems to a certain extent (Berry and Widder,2014).In addition, networked structure is closely related to ecosystem functions (Bastolla et al.,2009).Considering that bacteria and microeukaryotes significantly contribute to the biodiversity of ecosystems and play a key role in food webs and biogeochemical cycles (Fuhrman, 2009; Crits-Christoph et al., 2016), more data are needed on cross-domain networks between bacteria and microeukaryotes.Understanding and preserving this cross-domain microbial network structure is important for future ecosystem conservation.Recent advances in sequencing techniques and bioinformatics now enable an unprecedented level of investigation into complicated microbial ecological networks.Some findings have been obtained from single bacterial or microeukaryotic communities in soil (Chen et al.,2019a), reservoirs (Xue et al., 2018b), ocean (Milici et al., 2016), and the human body (Jackson et al.,2018).A global bacterial co-occurrence network has also been inferred from samples and sequence variants from 14 habitats in the Earth Microbiome Project dataset (Ma et al., 2020).All of these findings highlight the interconnection patterns of single microbial communities in single or various environments.In recent years, several studies have begun to focus on cross-domain microbial co-occurrence.For example, both bacterial and microeukaryotic networks are more stable and play more important roles in biogeochemical cycling in higher mean annual precipitation deserts than in lower (Feng et al., 2021).In a study of temperature gradient-related microbial population dynamics and network interactions, Anderson et al.(2021) found that increased eukaryote-eukaryote interactions under warm temperatures (e.g., Bacillariophyta-Dinophyceae) and the emerging contribution of bacteria to interactions and network connectivity under cold conditions.These studies contribute to our comprehension of microbial community organization in ecosystems, offering new perspectives on biogeochemistry modeling and ecosystem health assessment.However, ecological investigations of cross-domain microbial networks are still limited(Tan et al., 2015; Zancarini et al., 2017).
One fundamental question is whether it exists and what the relationship is between the complexity and stability of cross-domain microbial networks(Okuyama and Holland, 2008; Landi et al., 2018;Yuan et al., 2021).Previous studies have provided key information on the relationship between the complexity and stability of networks in a single domain.For example, Thébault and Fontaine (2010)have found that more complex architecture promotes stability in mutualistic eukaryotic networks, whereas the stability of trophic networks is enhanced in more simple architectures.In terms of bacterial networks,it has also been shown that the complexity of networks causes stability under warming conditions(Yuan et al., 2021).In contrast, studies on the relationship between complexity and stability in cross-domain microbial networks are still rare.
The second question refers to the relative importance of bacteria and microeukaryotes in constructing cross-domain microbial networks.Crossdomain co-occurrence of microorganisms is a common ecological phenomenon, as different microorganisms perform various ecological functions through forming a complex ecological interaction network (Bascompte,2009; Needham et al., 2017; Pande and Kost, 2017;Mikhailov et al., 2019).Shi et al.(2019) have found that archaea played a critical role in the construction of soil microbial co-occurrence network (including bacteria, fungi, and archaea) in the Qinghai-Xizang Plateau, because archaea not only had more connections than others in the network, but also greatly enhanced the robustness of the network.However, we know surprisingly little about the relative importance of bacteria and microeukaryotes for structuring their co-occurrence network in natural ecosystems.
The third question is the potential environmental drivers of cross-domain microbial networks and the differences in their effects on bacteria and microeukaryotes.Understanding the responses of microbial networks to environmental forcing features such as seasonality (Gilbert et al., 2012) and natural disturbance (Jones et al., 2008) is a central theme in microbial ecology, which can provide much valuable information.Specifically, it is possible to determine the general preferences of taxa for specific environmental factors when environmental parameters are included in the network (Faust and Raes, 2012; Vacher et al., 2016).For example, Wan et al.(2020) found significant differences in the environmental drivers of bacterial, fungal, and diazotrophic networks in paddy soils of eastern China.At the continental scale (ca.12 000 km),temperature, salinity, nitrite, nitrate nitrogen, total nitrogen, and grain size were important environmental factors driving the three different protozoans(diatoms, caudodonts, and ciliates) networks in intertidal sediments (Pan et al., 2020).In most papers addressing microbial networks, bacteria and microeukaryotes have been described separately.By contrast, we analyzed both components (bacteria and microeukaryotes) to provide a more comprehensive understanding of the response of cross-domain networks to environmental disturbances.
Sansha Bay is a typical subtropical bay located in southeastern China.In recent years, water quality and ecosystem of Sansha Bay have been strongly affected by land pollution, coastal industries,aquaculture, and urbanization, resulting in severe habitat degradation (Wu et al., 2012).The ecological condition of Sansha Bay is the epitome of many subtropical bays, and we expect the cross-domain microbial co-occurrence network in this bay to be a representative one.
In this study, water samples were collected from Sansha Bay during four seasons (January, April,July, and October of 2019).Both 18S rRNA and 16S rRNA genes were sequenced using high-throughput sequencing.We hypothesize that (1) there is no significant relationship between the complexity and stability of cross-domain networks under seasonal variation; (2) bacteria and microeukaryotes are equally important in the construction of crossdomain microbial networks in different seasons;(3) the same environmental factors drove the crossdomain networks in different seasons, and environmental factors had the same effects on bacteria and microeukaryotes components in the networks.
2 MATERIAL AND METHOD
2.1 Study area, sampling, and environmental factors
This study was conducted in Sansha Bay(119°30′E–120°5′E, 26°30′N–26°51′N), located in the northeastern area of Fujian Province, China, is connected to the East China Sea by only a narrow outlet (about 2.9 km wide).This subtropical bay was seasonally sampled at two different depths from surface to bottom waters (January, April, July, and October of 2019, Fig.1).A total of 120 water samples (30 samples were collected in each season)were collected using Niskin bottles (Supplementary Table S1).The samples were transported to the laboratory and processed immediately.Water samples were pre-filtered through a 200-μm sieve to minimize the interference of debris, mesoplankton, and macroplankton for community analyses.Then, a 1-L water sample was filtered through a 0.22-μm polycarbonate membrane (47 mm in diameter,Millipore, Billerica, MA, USA).The filter membranes were stored at -80 °C until DNA extraction.
Water temperature, salinity, and depth were measured in situ with conductivity-temperaturedepth (CTD) oceanic profilers (AML Base X).Other chemical parameters, including pH, total nitrogen(TN), dissolved inorganic nitrogen (DIN), nitrite(NO-2), nitrate (NO-3), ammonium (NH4+), phosphate(PO3-4), total phosphorus (TP), dissolved silicate(DSi), and chlorophylla(Chla) were also measured according to standard methods (Xie et al., 2020).
2.2 DNA extraction, PCR, and Illumina sequencing
DNA was extracted using the Fast DNA spin kit for soil (BIO101 systems, MP Biomedicals, Solon,OH, USA) following the manufacturer’s instructions.Bacterial DNA was amplified with tagged primers targeting the V3–V4 region of the 16S rRNA gene(341F, 5′-CCTAYGGGRBGCASCAG-3′; 806R, 5′-GGACTACNVGGGTWTCTAAT-3′) (Liu et al., 2019).The PCR mixture (30 μL) contained 15 μL of Phusion Master Mix (New England Biolabs, Beverly,MA, USA), 0.2 μmol/L of forward and reverse primers, and approximately 10-ng template DNA.PCR reactions included an initial denaturation at 94 °C for 5 min, followed by 30 cycles of 94 °C for 30 s, 57 °C for 30 s, 72 °C for 15 s, and a final extension at 72 °C for 10 min.The V9 region of the microeukaryotic 18S rRNA gene was amplified with primer pair 1380F (5′-CCCTGCCHTTTGTA CACAC-3′) and 1510R (5′-CCTTCYGCAGGTTC ACCTAC-3′) (Amaral-Zettler et al., 2009).PCR mixtures (30 μL) were prepared in triplicate and each contained 10 ng of DNA template.The PCR reaction was performed at 94 °C for 5 min, followed by 30 cycles at 94 °C for 30 s, 55 °C for 30 s, 72 °C for 15 s and a final cycle of 10 min at 72 °C.All triplicate PCR products (both 16S and 18S) were pooled in equal quantities and purified using GeneJET Gel Extraction Kit (Thermo Scientific, Hudson, NH,USA).Libraries were generated using the NEB Next Ultra DNA Library Prep Kit for Illumina (New England Biolabs, Beverly, MA, USA) following the manufacturer’s instructions, and barcode indexes were added.The library quality was assessed in the Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA).16S libraries were sequenced on the Illumina HiSeq2500 platform (Illumina Inc., San Diego,CA, USA) using a paired end strategy.18S libraries were sequenced on an Illumina X Ten platform (Illumina Inc., San Diego, CA, USA)using a paired-end (2×250 bp) approach.
2.3 Bioinformatics
Paired-end Illumina V9 region of 18S rRNA gene sequences and V3–V4 region of 16S rRNA gene sequences were processed using Vsearch 1.9.1(Rognes et al., 2016).To denoise sequences and remove chimeras, the unoise3 algorithm with default settings (minsize=8) was used before the downstream analyses.Quality-filtered reads were then clustered into operational taxonomic units(OTUs) at a cutoff of 97% for both 16S rDNA and 18S rDNA sequences.Representative sequences from each OTU were aligned against the Protist Ribosomal Reference database (PR2 version 4.7.2)(Guillou et al., 2013) for microeukaryotes and SILVA database (version 132 NR) (Quast et al.,2013) for bacteria, respectively.Unassigned microeukaryotic OTUs (sequence similarity to a reference sequence is <80%) and microeukaryotic singletons (OTUs with only one sequence) were discarded before the downstream analyses.For the 16S OTUs, eukaryotic, chloroplast, archaeal,singletons, and unclassified sequences were removed before further analysis.Finally, to minimize biases associated with sequencing coverage, both 16S rDNA and 18S rDNA sequence data were normalized to the lowest number of sequences per sample, i.e., 39 206 sequences per sample.
2.4 Network construction
All cross-domain co-occurrence networks were constructed using the Molecular Ecological Network Analyses Pipeline (MENAP) (Deng et al., 2012).First, OTUs that occurred in more than half (>15 each season) of samples were used to construct the networks.Secondly, the Pearson’s correlation coefficient was calculated based on the logtransformed OTUs abundances.Finally, a random matrix theory (RMT)-based approach was utilized to automatically determine the correlation cut-off threshold, leading to the construction of highly reliable microbial ecological networks (Luo et al.,2007; Zhou et al., 2010).The greatest advantage of RMT-based network approach was the use of mathematically defined non-arbitrary correlation cutoffs, significantly reducing uncertainty in network construction and comparison (Yuan et al., 2021),and this method has been widely applied (Deng et al., 2016; Ma et al., 2016).Four seasonal crossdomain networks (one network per season) were constructed using data from each season, and the similarity threshold of 0.86 was determined by the RMT-based approach.For each observed network in this study, 100 corresponding random networks(identical number of nodes and edges) were constructed in the MENAP to test the significance of the observed networks (Maslov and Sneppen, 2002).Modular analysis was carried out using MENAP.All networks were visualized by the Gephi version 0.9.2 (Bastian et al., 2009).
2.5 Network analysis
To evaluate the difference in the complexity of the four seasonal cross-domain networks, network complexity indices such as nodes, links, average clustering coefficient, relative modularity, number of modules, and R square of power law were calculated in the MENAP.To further understand the network organizational principles, Spearman’s rank correlations were used to determine the relationships between various network topological indices and RM (how modular a network is as compared with the average expected modularity) (Thébault and Fontaine, 2010).Then, we calculated robustness (as expressed by natural connectivity) based on the“igraph” package of R v.4.0.2 to characterize the stability of the networks (Peng and Wu, 2016).Correlation analysis and Pearson’s correlation coefficient were used to identify potential relationship between the network complexity and stability indices.To determine the relative importance of bacteria and microeukaryotes in the construction of four seasonal cross-domain networks, specific taxa from the networks were removed one by one, and the robustness of remaining networks was calculated(Shi et al., 2019).To distinct the roles of each node in the cross-domain networks and further verify the relative importance of bacteria and microeukaryotes;Zi(within-module connectivity) andPi(amongmodule connectivity) values were calculated using MENAP.Nodes were defined as network hubs (Zi>2.5;Pi>0.6), module hubs (Zi>2.5;Pi≤0.6), connectors(Zi≤2.5;Pi>0.6) or peripherals (Zi≤2.5;Pi≤0.6)(Olesen et al., 2007; Deng et al., 2012).Among these, network hubs, module hubs, and connectors have been proposed to be kCystone taxa (Deng et al., 2012; Banerjee et al., 2018).Pearson’s correlation analysis was carried out using SPSS 20.0 to assess the relationships between the relative abundance of keystone taxa and environmental factors.To decipher the environmental drivers of networks and further investigate the ecological associations between species (microeukaryotes and bacteria) and environmental factors, all environmental factors measured in our study were included in the constructed cross-domain networks.Only connections that were strong (Pearson’s |R|>0.60) and statistically significant (P<0.01) were considered in this analysis.Then the directly linked species-environment connections were selected and visualized by Gehphi version 0.9.2.
3 RESULT
3.1 Microbial diversity and community compositions
For both the 18S and 16S rDNA datasets, a total of 4 704 720 high-quality sequences were recovered from 120 water samples (39 206 sequences per sample), respectively.For the 18S rDNA dataset, we finally detected 10 291 OTUs at 97% sequence similarity threshold, and the number of OTUs per sample varied from 536 (sample ES10-11D) to 2 023(sample ES4-2D).18 668 OTUs were obtained from the 16S rDNA dataset at the cutoff of 97% sequence identity, and the number of OTUs varied from 1 883(sample PS1-19B) to 5 898 (sample PS7-13D).Results from the diversity indices (ACE, Chao1,Shannon, and Pielou) demonstrated that April had the highest diversity for microeukaryotes, while bacterial diversity was higher in July (Supplementary Fig.S1).
The bacterial and microeukaryotic sequences were classified into different taxa levels, and the relative abundances of the top 20 most abundant groups in bacterial and microeukaryotic samples from different seasons were shown in Supplementary Table S2.The results indicated that the relative abundance proportions of dominant taxa differed significantly among various seasons.For microeukaryotic community, the relative abundance of Unclassified, Alveolate, and MAST was highest in January; April’s samples possessed a higher abundance of Labyrinthulea than other samples; high relative abundances of Alveolate,Chrysophyceae, and Mollusca were observed in the samples of July; and Dinoflagellata, Apicomplexa,and Alveolate were more abundant in October.For the bacterial community, Alphaproteobacteria,Gammaproteobacteria, Bacteroidetes, and Actinobacteria were the four most abundant bacterial taxa in each season.The sequence abundance proportion of Gammaproteobacteria was significantly higher in January than in other seasons, the proportion of Cyanobacteria was higher in January.
3.2 Linkage between cross-domain network complexity and stability
Four seasonal cross-domain networks were constructed, with the networks for January, April,July, and October containing 277, 455, 505, 185 nodes and 974, 1 103, 1 886, 355 edges, respectively(Fig.2).All observed networks exhibited scale-free characteristics (power-law:R2=0.829 for January network,R2=0.892 for April network,R2=0.824 for July network, andR2=0.886 for October network,respectively), which were different from their corresponding random networks (Erdos-Rényi model),indicating that the observed networks were nonrandom and had biological significance.In addition,all four networks exhibited modular structure(modularity>0.4 in all networks, Supplementary Table S3).
To examine if networks complexity were different among four seasons, changes in various network topological parameters were analyzed (Supplementary Fig.S2).Network size (total nodes) and connectivity(total links) strongly varied over time, they were low in January and October, but relatively high in April and July.Average clustering coefficient,average path distance, harmonic geodesic distance,relative modularity, number of modules, number of nodes in the largest module and efficiency all showed a similar trend.These results indicated that the network complexity changed significantly with the seasons, and the network complexity in January and October was lower than that in April and July.Moreover, the proportion of positive connections was dominant in each seasonal network (85.01% for January, 99.55% for April, 99.63% for July, and 97.91% for October, respectively; Fig.2), and the change trend was consistent with above complexity indicators.These results suggest that cooperative behaviors were predominant in the cross-domain interactions between bacteria and microeukaryotes and varied seasonally.As the proportion of positive connections increased, the relative modularity of networks increased significantly (P<0.01, Spearman),which indicated that the underlying cooperative behaviors further affected the organizational principles of the networks (Fig.3a).

Fig.2 Succession of bacterial and microeukaryotic cross-domain networks over seasons
Robustness (natural connectivity) was used in our study as an index of stability, and the value decreased from 0.078 to 0.030 (Fig.3b).This suggested that the cross-domain network was more stable in January and the stability then decreased.More importantly, significant correlations were observed between network stability and network complexity.The proportion of positive connections was negatively correlated with network robustness(Pearson’sR=-0.973,P<0.01).Similarly, significant negative correlations were observed between modularity and network robustness (Pearson’sR=-0.994,P<0.01).However, centralization of degree was positively correlated with network robustness(Pearson’sR=0.998,P<0.01).Collectively, these results indicated that more frequent cooperation and more complex modular structures weaken the stability of cross-domain networks.
3.3 Relative importance of bacteria and microeukaryotes in cross-domain networks

Fig.3 Relationship between the network complexity and network stability
To reveal the relative importance of bacteria and microeukaryotes in the cross-domain networks,analyses about nodes and edges were conducted.Most of the nodes (OTUs) in four cross-domain networks were affiliated to bacteria, and the proportion varied in season (82.31% for January, 73.41% for April, 71.49% for July, and 85.41% for October,respectively; Fig.2).Taxonomic relatedness was a critical factor in determining the modular structure in the network.For instance, in all networks,microeukaryotic and bacterial nodes formed significantly different modules respectively.The networks from January, April, and July mainly harbored nodes belonging to Gammaproteobacteria,Alphaproteobacteria, Bacteroidetes, Actinobacteria,and Deltaproteobacteria.In October, the network occupied nodes mainly assigned to Alphaproteobacteria,Actinobacteria, Gammaproteobacteria, Cyanobacteria,and Chloroflexi (Supplementary Table S4).Moreover,only 12 nodes were shared among four networks;most of the nodes were unique to each seasonal network, which suggested that most microeukaryotic and bacterial groups had their special niches in different seasons (Supplementary Fig.S3).
When considering all edges (connections) in the four networks.Bacteria-bacteria connections were dominated in all networks, followed by microeukaryotesmicroeukaryotes connections, and the number of microeukaryotes-bacteria connections was relatively low (Supplementary Fig.S4).The percentage of positive connections was generally higher than the negative ones in any type of connections(Supplementary Fig.S4).We further calculated the connections information between different taxa in different seasons to predict possible interactions.These results show that connections in the four seasonal networks were concentrated mainly among abundant (number of nodes) bacterial taxa (Supplementary Figs.S5–S8).In January,there were more connections observed among Gammaproteobacteria, Alphaproteobacteria,Bacteroidetes, and Actinobacteria (Supplementary Fig.S5).In April, the connections associated with Gammaproteobacteria, Alphaproteobacteria,Actinobacteria, Bacteroidetes, and Deltaproteobacteria were dominant (Supplementary Fig.S6).In addition to some dominant taxa (Gammaproteobacteria,Alphaproteobacteria, Bacteroidetes, Deltaproteobacteria,and Actinobacteria), Syndiniales, Chloroflexi,Bacillariophyta, and Cyanobacteria also had more related connections in July (Supplementary Fig.S7).In October, Gammaproteobacteria, Alphaproteobacteria,Bacteroidetes, Chloroflexi, and Actinobacteria had a high proportion of connections (Supplementary Fig.S8).These results suggest that bacteria OTUs were the main contributors to the complexity of cross-domain networks, and always occupied a key position in cross-domain interactions, which is completely different from the hypothesis that bacteria and microeukaryotes are equally important in the construction of cross-domain microbial networks in different seasons.
To verify the relative importance of bacteria and microeukaryotes in the cross-domain networks, we simulated random species extinction and calculated the robustness for complete cross-domain networks,bacteria-absent networks, and microeukaryotesabsent networks (Fig.4).The robustness of all networks, except for bacteria-absent networks,gradually decreased as more nodes of OTUs were removed.That is, the robustness change trend of the complete network was more similar to the microeukaryotes-absent networks, while was significantly different from the bacteria-absent networks.This was a good illustration of the key role of bacteria in cross-domain networks.To further identify important bacterial or microeukaryotic taxa in the cross-domain networks, we removed specific taxa in the networks one by one, simulated random species extinction, and calculated the robustness(Fig.5).In January, when Alphaproteobacteria or Gammaproteobacteria were removed, the trend of robustness was significantly different from the complete network.This result indicates that Alphaproteobacteria and Gammaproteobacteria were important in the construction of the January network.Similarly,Prymnesiophyceae and Gammaproteobacteria played important roles in the construction of the April network.In July, Alphaproteobacteria,Gammaproteobacteria, Bacteroidetes, and Actinobacteria were important taxa in constructing the network.As for October, Alphaproteobacteria,Gammaproteobacteria, Chloroflexi, Bacteroidetes,and Actinobacteria were more important in network construction.These results show that bacteria were the most important contributor to the stability of the cross-domain networks.
3.4 Keystone OTUs of four seasonal cross-domain networks
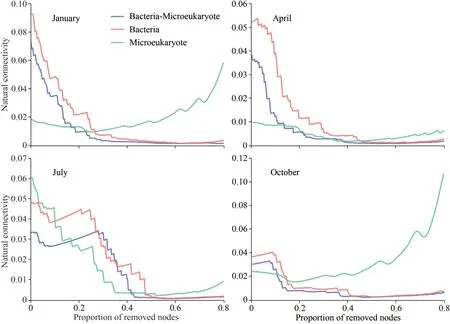
Fig.4 Network robustness analysis of containing bacteria, microeukaryote, and bacteria-microeukaryote microbial groups in Sansha Bay
Keystone OTUs (module hubs, network hubs,and connectors) were identified based onZiandPiof networks (Fig.6a).Most of the nodes in each network were peripherals (99.64% for January,98.90% for April, 98.81% for July, and 100% for October, respectively).No network hubs and connectors were found in any of the networks across all seasons.We therefore investigated mainly the module hubs of networks.Three module hubs were found in the January network, and they all belonged to Rhodobacteraceae (Supplementary Table S5).Five module hubs were present in the April network,including one Plagioselmis OTU, one Woeseiaceae OTU, one Rhodobacteraceae OTU, and two Actinomarinales OTUs (Supplementary Table S5).A total of six module hubs were detected in the July network, including one Cryptomonadales OTU, one Syndiniales OTU, two SAR11_clade OTUs, one Actinomarinales OTU, and one Gammaproteobacteria OTU (Supplementary Table S5).No module hub was found in the October network.Additionally,module hubs had no overlap between different networks, showing that bacterial or microeukaryotic species responsible for these topological roles varied across different seasons.
Keystone OTUs (module hubs) were correlated with measured environmental factors to investigate their relationships (Fig.6b).The Keystone OTUs showed the strongest species-environment relationships in July network, followed by the January network, and the weakest relationships were observed in April network.In January, all three bacterial keystone OTUs showed significant positive correlations with salinity, while two of them were significantly negatively correlated with temperature and nutrients (such as PO4and NH4).In April network, depth and temperature were the two most important environmental filters shaping the bacterial keystone OTUs.In July, bacterial keystone OTUs presented stronger species-environment relationships than microeukaryotic keystone OTUs.On the one hand, these results showed the importance of bacterial OTUs in the cross-domain networks; on the other hand, they also revealed the relationship between networks and environmental variables.
3.5 Environmental driver of seasonal crossdomain networks
In addition to investigating the relationship between environmental variables and keystones OTUs, we incorporated environmental factors into the constructed cross-domain networks to decipher the environmental drivers of networks more comprehensively (Fig.7).All selected speciesenvironment networks suggested that environmental factors had relatively strong correlations with bacterial taxa in every season.Co-occurrence of bacterial and microeukaryotic taxa in January was primarily constrained by temperature and salinity.In April, depth and temperature emerged as the most important impact on the cross-domain network compared with other factors.In July, the crossdomain network was mainly affected by depth,temperature, and salinity, but DSi, NO2, DIN, NO3,TN, and TP also played an important Roles.Depth was the most important factor affecting the crossdomain network in October, followed by salinity,NO2, and Chla.
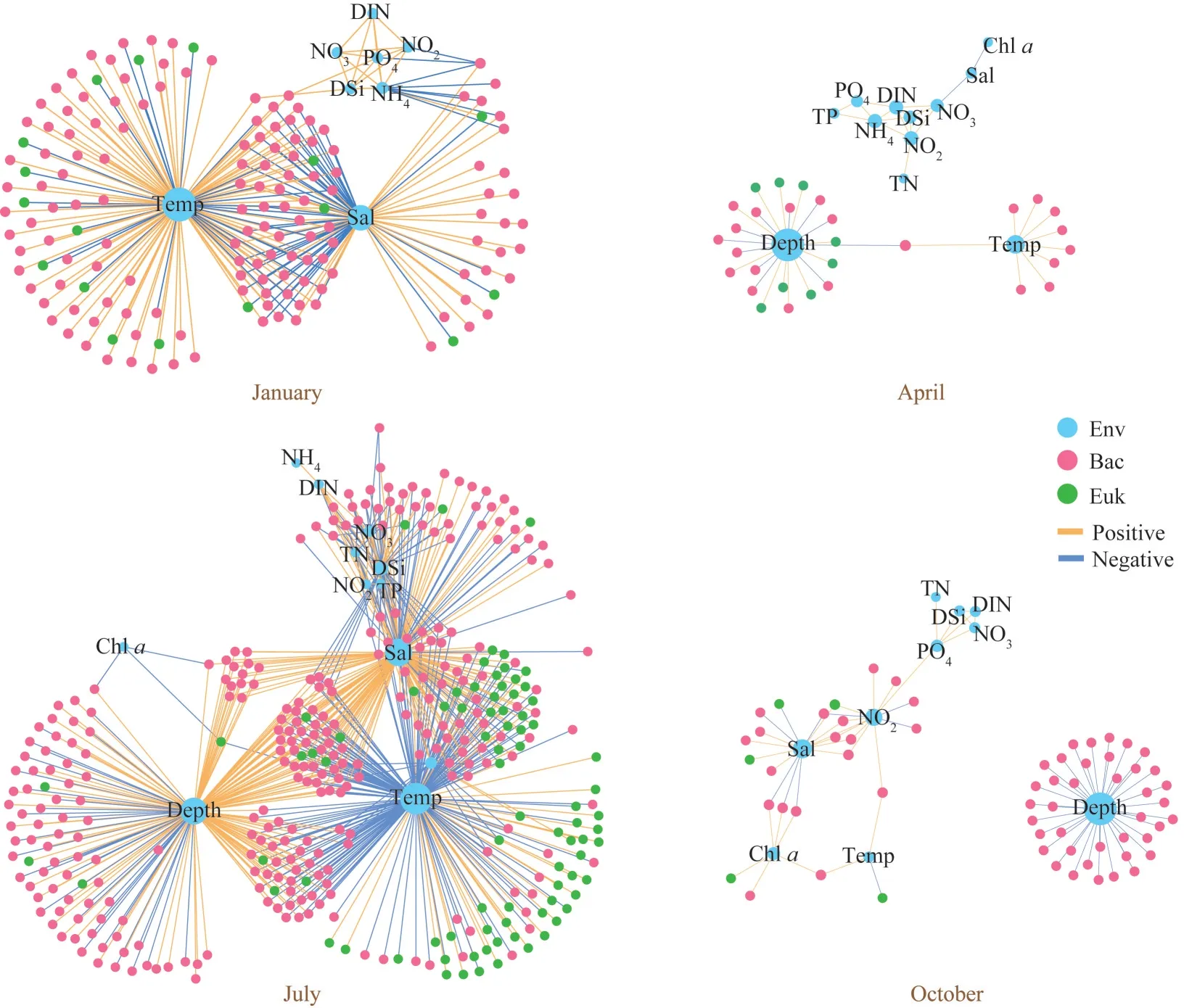
Fig.7 Visualization of directly linked species-environment connections in the four cross-domain networks
We further analyzed the differences in the effects of environmental factors on bacteria and microeukaryotes in the network (Supplementary Fig.S9).In all networks except for April, we found that environmental factors had the most connections with Alphaproteobacteria, Gammaproteobacteria,Bacteroidetes, Actinobacteria, and Deltaproteobacteria.Futhermore, temperature and salinity were the main factors that affected the microeukaryotes nodes in these networks.In the April network, salinity had strong positive relationships with microeukaryotes,and strong negative relationships with bacteria.
4 DISCUSSION
4.1 Increasing of positive connections and modularity weakened cross-domain network stability
Significant negative correlations were observed between proportions of positive connections and network robustness in this study (Fig.3b).In other words, the increase in the proportion of cooperation within microorganisms might reduce the stability of cross-domain networks.Studies of interaction networks have contributed to our understanding of how interactions are related to ecological stability of microbiome communities (Neutel et al., 2002;Thébault and Fontaine, 2010).The reason for this might be that cooperation caused coupling between species and positive feedback, as cooperation could create dependency and the potential for mutual downfall.For example, the decrease of one species in abundance pulled others down with it and destabilized the system (Coyte et al., 2015).Interestingly, an analysis of macroscopic communities predicted that ecological stability could be maximized under conditions of moderately frequent cooperative interactions (Mougi and Kondoh, 2012).However, all of the networks in our study had highly frequent cooperative interactions.Therefore, the results should be interpreted with more caution,namely, based on highly frequent cooperative interactions, the continued increase in cooperative interactions would reduce the stability of crossdomain microbial networks.
The modularity of the cross-domain network was lower in January, but higher in April, July, and October, the centralization of the degree of crossdomain network showed the opposite trend.Previous studies have interpreted modules as functional units(Luo et al., 2006) or niches (Eiler et al., 2012) in the microbial communities.Modularity represented the extent to which a network can be divided into modules, and could potentially affect network robustness (Olesen et al., 2007).For example, some studies had shown that whether in macroecology or microecology, network stability increased with network modularity (Montoya et al., 2006; Yuan et al., 2021).Researchers have also proposed a reasonable explanation that modularity promoted community stability by buffering the spread of a perturbation across the entire network (May, 1972;Gainsbury and Meiri, 2017).However, our study showed the opposite result that increased modularity weakened the stability of the network (Fig.3b),which supported the argument of some authors(Pimm, 1979; Solow et al., 1999).These studies further complicated the relationship between modules and stability.Considering that the indirect influence of the environment on network stability through modularity was common (Liu et al., 2021),we would fully explore the role of environmental factors in future studies.For example, samples were split into groups according to a key variable such as water depth or health status, enabling the construction of separate networks for each sample group to provide a more comprehensive explanation of the relationship between modularity and stability(Faust, 2021).
In summary, by examining the temporal crossdomain network dynamics of microbial (bacteria and microeukaryotes) communities in response to seasonal change, this study provided important insights into the relationships between microbial cross-domain network complexity and stability.
4.2 Bacteria exhibited a pivotal role in the crossdomain network
In terms of connections in the four networks, we found that species within bacteria (intra-domain)built the most connections, followed by species within the microeukaryotes (intra-domain), and the connections between bacteria and microeukaryotes(inter-domain) were the least (Supplementary Fig.S4).In addition, microorganisms within one domain mostly correlated positively with each other(Supplementary Fig.S4).These results suggested that OTUs within the domain, especially in the bacterial domain, could more easily form a mutualistic community.Considering that microorganisms are highly dependent on cross-feeding, co-aggregation,co-colonization, and niche overlap (Kylafis and Loreau, 2011; Faust and Raes, 2012), it was not surprising to observe such positive connections in natural ecosystems (Faust et al., 2015).Millions of tons of feed were required annually to support the production ofLarimichthyscroceain Sansha Bay, of which 5%–10% was not utilized and decomposed into organic and inorganic matter in the water column (Duan et al., 2001) and eventually led to eutrophication (Chen et al., 2019b).Therefore, the abundant nutrients caused by aquaculture activities in Sansha Bay might weaken the competitive relationships within the microorganisms and ultimately produced the current results (Mikhailov et al., 2019).
Bacteria formed greater connection numbers in the network compared with those of microeukaryotes(Supplementary Fig.S4), which could explain why omitting bacteria dramatically affected the natural connectivity of the cross-domain microbial networks in our study (Fig.4).Additionally, we observed that connections between Gammaproteobacteria,Alphaproteobacteria, Bacteroidetes, and Actinobacteria were abundant in the four networks (Supplementary Figs.S5–S8).Consistent with these results, the absence of some or all taxa (Gammaproteobacteria,Alphaproteobacteria, Bacteroidetes, and Actinobacteria)had significant effects on the natural connectivity of cross-domain networks in different seasons (Fig.5).A previous study of global ocean sampling metagenomic data showed the widespread types of bacterial type Ⅵand Ⅳ secretion systems (T6 SS and T4SS) in Gammaproteobacteria and Alphaproteobacteria (Persson et al., 2009).In addition,Alphaproteobacteria and Gammaproteobacteria have been reported to participate in the balance of the global nitrogen budget via nitrogen fixation or denitrification (Tsoy et al., 2016).Members of Actinobacteria contributed to heterotrophic nitrification and played a key role in nutrient and energy cycling in aquatic habits (Elifantz et al.,2005).The highly diverse phylum Bacteroidetes can efficiently degrade many high-molecular-weight compounds, such as proteins, cellulose, pectin, and chitin (Xue et al., 2018a).Therefore, it was reasonable to infer that more intra-domain bacterial connections would further promote the carbon and nitrogen cycles and maintain seasonal stability of ecological functions in this subtropical aquaculture bay.
In this study, all keystone OTUs from the four cross-domain networks were related to module hub functions, and most of them belonged to bacteria(Fig.6a).These keystone OTUs were closely related to the OTUs within the module, therefore they might play a predominant role in determining the structure of the whole community (Zhou et al.,2011).Marine Rhodobacteraceae were key players of biogeochemical cycling, comprised up to 30% of bacterial communities in pelagic environments and were often mutualists of eukaryotes (Simon et al.,2017).Plagioselmisprolongawas free-swimming and highly tolerant of low salinity.Previous studies had found thatP.prolongawas present throughout the year in some coastal waters, and the peak abundance was usually detected in the spring(Cerino and Zingone, 2006; Xing et al., 2008).Metagenomes of Woeseiaceae encoded an incomplete denitrification pathway, which included genes for NO-2reduction (nirS) and NO reduction(norB) to the ozone-depleting greenhouse gas N2O(Hinger et al., 2019).Actinomarinales were the only known exclusively marine, free-living, planktonic Actinobacteria, and represented an important player in the microbial ecology of the oligotrophic ocean(Ghai et al., 2013; López-Pérez et al., 2020).SAR11 is an ancient and diverse clade of heterotrophic bacteria that dominates ocean surface bacterioplankton communities and played an important role in the ocean carbon cycle (Grote et al., 2012).Syndiniales are widespread in marine environments (Chambouvet et al., 2011; Clarke et al., 2019), where they can infect several types of plankton (Skovgaard, 2014).Therefore, they are also key players in the structure and connectivity of plankton food webs (Zamora-Terol et al., 2021).These results again suggest the importance of bacterial taxa in constructing the cross-domain networks and maintaining the ecological function.
4.3 Relationship between the cross-domain networks and environmental variables
Previous studies have shown that the presence of keystone species in the microbiomes did not certainly guarantee their influence, as many factors might still determine their distribution and efficacy(Banerjee et al., 2018).Therefore, understanding the keystone species in the network and the environmental variables that might influence can better guide the manipulations of microbial communities in natural ecosystems (Toju et al.,2018).In our study, the environmental variables linked to keystone OTUs greatly differed between seasons, reflecting that the keystone OTUs of crossdomain network had different ecological niches between seasons (Fig.6b).Moreover, dissimilar environmental drivers of seasonal cross-domain networks were also found (Fig.7).Temperature and salinity were the best explanatory factors predicting the cross-domain network of January.Depth and temperature were the best explanatory factors predicting the cross-domain network of April.Temperature, salinity, and depth were the best explanatory factors predicting the cross-domain network of July.Depth, salinity, and NO2were the best explanatory factors predicting the cross-domain network of October.This discrepancy might be related to the environmental heterogeneity between seasons (Horner-Devine et al., 2004).Temperature is one of the most important predictors of aquatic bacterial and microeuakryotic co-occurrence networks(Ren et al., 2019; Zhu et al., 2019; Pan et al., 2020).In our study, the temperature had the majority connections with bacterial nodes in January’s networks (cold networks), and a large number of connections with both microeukaryotic and bacterial nodes in the July network (warm network).Interestingly, Anderson et al.(2021) found that interactions among 18S groups were more common in the warm microbial network in Skidaway River estuary (a subtropical estuary), whereas in the cold network, although interactions among 18S groups remained common, 16S groups contributed more to the top interactions.This might provide an explanation for our results that water temperature correlates with several abiotic and biotic factors(Wang et al., 2019), and thus exert a strong control on seasonal cross-domain networks in this bay.As a major environmental variable across many ecosystems, salinity has been reported to drive microbial community assembly strongly (Zhang et al., 2017; Mo et al., 2018; Santoferrara et al., 2018).Therefore, salinity might play a pivotal role in regulating seasonal cross-domain networks.Additionally, depth was one of the most important predictors of the cross-domain networks in April,July, and October.A reasonable explanation for this was that the strong vertical exchange of the water column in January led to a lower temperature difference between the surface and bottom water,while the weaker vertical exchange of the water column in other seasons led to a higher temperature difference between the surface and bottom water.
In addition to understanding the environmental drivers of cross-domain networks, we could also identify the different effects of these environmental factors on the bacterial and microeukaryotic taxa in the network (Supplementary Fig.S9).The stronger correlations between bacterial taxa and environmental factors in each season indicated that environmental change was mainly driving the cross-domain network through bacterial taxa, which again confirmed the importance of bacteria in the construction of crossdomain network.Interestingly, the bacteria taxa that were more influenced by environmental factors were almost identical to those that played an important role in network construction (i.e., OTUs that were the important contributors to the network complexity and network stability; Fig.5, Supplementary Figs.S5–S8, and Supplementary Table S4).Therefore,we believe that environmental factors could further drive the cross-domain co-occurrence of microorganisms by influencing Gammaproteobacteria,Alphaproteobacteria, Bacteroidetes, Actinobacteria,and Deltaproteobacteria.
5 CONCLUSION
This study provides important insights into the seasonal changes of cross-domain microbial networks in a typical subtropical bay.Significant correlation was observed between network complexity and network stability, in which more frequent cooperative and more complex modular structures weaken the stability of cross-domain networks.Compared with microeukaryotes, bacteria occupied more nodes and keystone OTUs, built more connections, and were the most important contributors to network complexity.Moreover, the differences of natural connectivity between bacteriaabsent networks and complete networks were larger than that between microeukaryote-absent networks and complete networks.These results indicate that bacteria were key for constructing the cross-domain networks and maintaining the network stability,with Gammaproteobacteria, Alphaproteobacteria,Bacteroidetes, and Actinobacteria as representative taxa.Environmental variables affected the cooccurrence patterns (including network complexity,network stability, and the relationships between them) of cross-domain networks, and this process was mainly achieved by affecting bacterial taxa(e.g., Gammaproteobacteria, Alphaproteobacteria,Bacteroidetes, Actinobacteria, and Deltaproteobacteria).Therefore, the importance of bacterial communities and some specific bacteria taxa should be fully recognized in future cross-domain network conservation.
6 DATA AVAILABILITY STATEMENT
16S rRNA gene sequences have been submitted to the NCBI Sequence Read Archive (SRA) database under the BioProject number PRJNA747131 and the accession number SRP328863.18S rRNA gene sequences have been submitted to the NCBI Sequence Read Archive (SRA) database under the BioProject number PRJNA747143 and the accession number SRP328880.
7 ACKNOWLEDGMENT
The authors would like to thank Dr.Luciana SANTOFERRARA (Hofstra University, USA) for suggestions to improve the writing, Dr.Yuanyuan MO (Institute of Urban Environment, Chinese Academy of Sciences, China) for constructive comments on the manuscript, and Prof.Jianyu HU(Xiamen University, China) for providing CTD data.We thank Dr.Peng XIAO (Institute of Urban Environment, Chinese Academy of Sciences, China)for the data processing.
 Journal of Oceanology and Limnology2023年6期
Journal of Oceanology and Limnology2023年6期
- Journal of Oceanology and Limnology的其它文章
- Trends of carbon and nutrient accumulation through time in the Andong salt marsh, Hangzhou Bay, China*
- Physical processes determining the distribution patterns of Nemopilema nomurai in the East China Sea*
- Comparison in structure and predicted function of epiphytic bacteria on Neopyropia yezoensis and Neopyropia katadae*
- Interaction between macroalgae and microplastics: Caulerpa lentillifera and Gracilaria tenuistipitata as microplastic bio-elimination vectors*
- Lake regime shift from submerged macrophyte to phytoplankton affected phosphorus speciation in sediment and eutrophic state in Caohai Lake, Guizhou, China*
- Temporal characteristics of algae-denitrifying bacteria co-occurrence patterns and denitrifier assembly in epiphytic biofilms on submerged macrophytes in Caohai Lake, SW China*