
The Status Quo and Enlightenment of the Foreign Extended Clinical Trial System
2023-12-23 09:16:18TengXinyiTianLijuan
亚洲社会药学杂志 2023年4期
Teng Xinyi,Tian Lijuan
(School of Business Administration,Shenyang Pharmaceutical University,Shenyang 110016,China)
Abstract Objective To study the extended clinical trial systems in the United States,the European Union and Australia,and to provide a reference for the improvement of China’s extended clinical trial system.Methods Literature research method,risk management analysis method and comparative research were used in this paper to analyze the development history,scope of use,status quo of the extended clinical trial systems in the United States,the European Union,and Australia.Then,the current situation and shortcomings of China’s extended clinical trial system were compare with these foreign countries so as to put forward some suggestions for improvement.Results and Conclusion China should improve existing laws and regulations by establishing a fast application procedure,increasing application channels,and providing an information disclosure platform to meet the needs of patients in extended clinical trials.
Keywords: extended clinical trial regime;application process;development history
Extended clinical trials,also known as compassionate medications,are defined by the US Food and Drug Administration as measures in which patients with an immediately life-threatening or serious illness acquire an investigational new drug(IND) for treatment outside of clinical trials when there are no other alternative therapies available[1].
At present,the United States,the European Union,Australia and other countries and regions have established a legal system for extended clinical trials.According to FDA data,the number of applications for extended clinical trials in the United States has continued to grow in recent years,and the approval rate of applications is greater than 95%[2].More than 20 member states in the European Union have established extended clinical trial regulations and related application processes[3].Australia has more than 60 000 applications for extended clinical trials each year[4].While these needs for compassionate medication support research on rare diseases,different countries also should establish and improve extended clinical trial systems as soon as possible to ensure the safety of patient medication.
In 2017,China issued the “Opinions on Deepening the Reform of the Review and Approval System to Encourage the Innovation of Pharmaceutical Medical Devices”,which for the first time proposed to carry out extended clinical trials at the regulatory level.The “Regulations on the Administration of Extended Clinical Trials of Medical Devices(Trial)” issued in 2020 systematically put forward the responsibilities of extended clinical trials,the supervision of the whole process,and data collection and analysis for relevant parties.However,at present,China’s extended clinical trial system has not yet been perfected.Therefore,to ensure the drug demand of patients to the greatest extent based on balancing risks and benefits is an urgent problem that needs to be studied.
1 Foreign extended clinical trial system
1.1 Extended clinical trial system in the USA
The United States is the first country to establish an extended clinical trial system.In the early stages of its implementation,although the FDA took measures to give patients the right to use extended drugs,it lacked specific regulatory support.With the increase in the demand for clinical drugs,a specific system has been gradually formed for compassionate medication.In the United States,compassion medicines are currently used in a variety of different medical specialties,including but not limited to oncology,hematology,infectious diseases,gastroenterology,transplantation,and ophthalmology[5].
1.1.1 Regulatory documents related to compassionate medication in the USA
Over the years,the FDA has issued and revised a number of laws,and it has gradually refined and improved the applicable standards for compassionate medication,application conditions,patient types,and the rights and obligations of relevant subjects,as detailed in Table 1.

Table 1 Regulatory documents related to compassionate medication in the USA
1.1.2 Scope of application of compassionate medication in the United States
In the United States,compassionate medications are primarily suitable for: (1) Serious or immediately life-threatening diseases,but no suitable therapy;(2)The potential benefits and potential risks are justified;(3) There is no threat to the initiation,implementation,or completion of marketing-approved clinical trials in support of expanded access (in principle,expanded access is a pathway for patients who are unable to enroll in clinical trials).
1.1.3 Application category and approval process
According to the number of subjects,there are 3 types: Single patient,medium number of patients,and large number of patients.The increase in the number of patients has also brought more stringent conditions and a narrower scope of application.Individual patient requires patient-specific analysis and risk anticipation,so applications can be made at various stages.For a moderate number of patients,the sponsor must have early evidence of safety and efficacy to support its use.A large number of subjects means that the drug can be applied to a large population after the FDA has obtained sufficient evidence of safety and efficacy in phase II/III.
1.1.4 Principal responsibilities
In the process of implementing compassionate medication,the responsibilities of each subject are as follows.
(1) The patient consults the practicing doctor first,and then he/she tries and chooses his diagnosis and treatment method.
(2) Licensed pharmacists agree to participate in and monitor the patient’s nursing process,negotiate with pharmaceutical companies,determine the category of compassionate drugs they apply for in accordance with regulations.Then they submit written requests to the FDA and the Institutional Review Board (IRB),and promptly detect and report patient adverse events.
(3) Enterprises provide drugs and corresponding clinical trial programs according to their wishes,allowing the FDA to use them in the form of authorization,and providing necessary clinical trial drug information.
(4) The informed consent form is reviewed by the IRB,the FDA reviews the compassionate medication application,and the FDA approves the compassionate medication before it can be used.
1.1.5 Application and approval status of compassionate medication
In recent years,FDA has received thousands of applications for compassionate medication,onetenth of which are applications approved after revision,including patient safety evaluation and trial drug dosage.The approval status of various types of extended clinical trials in the United States from 2015 to 2019 is as follows (Table 2).
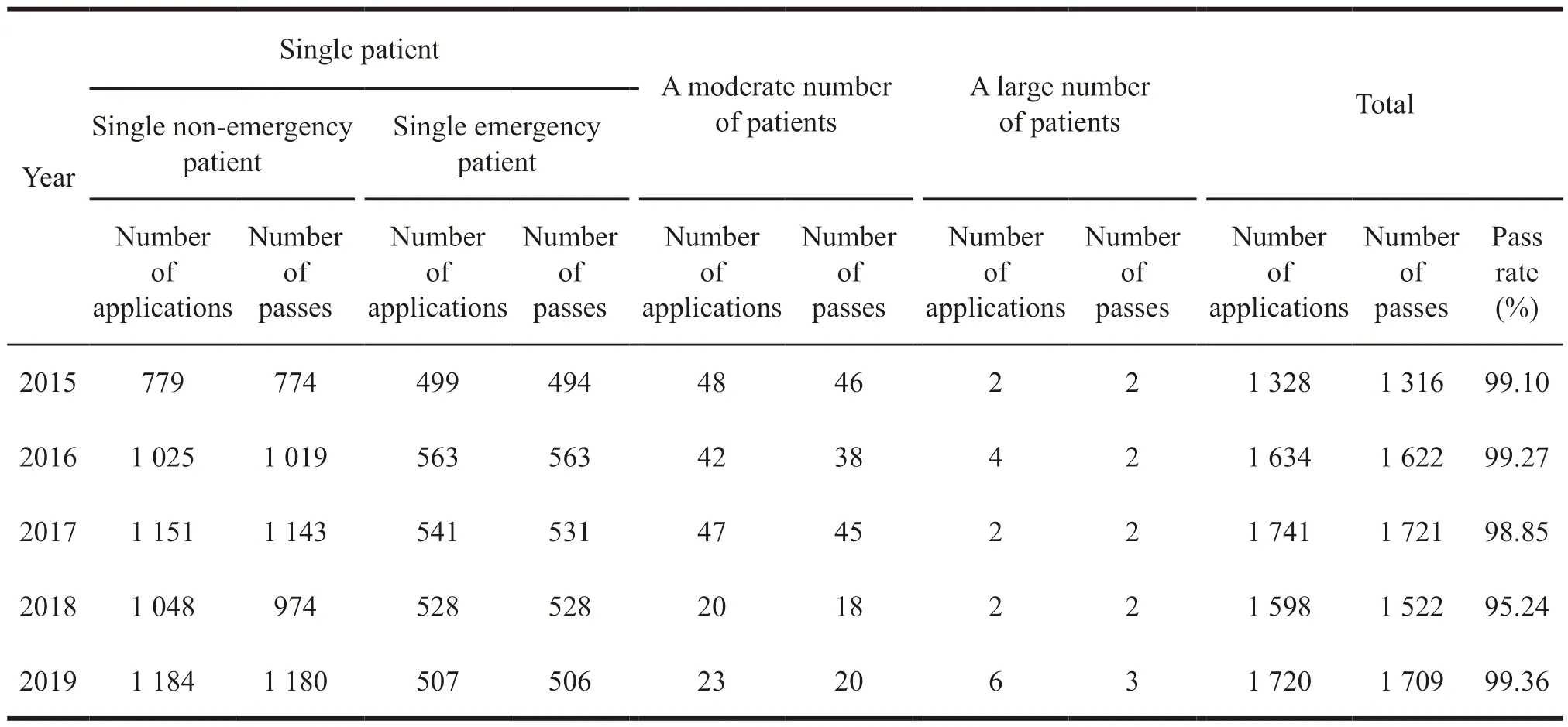
Table 2 Approval of class 3 extended clinical trials in the United States from 2015 to 2019
From Table 2,it can be found that the application and approval rate of various types of extended clinical trials is high,especially in a large number of patients categories,and the approval rate is close to 100%.the largest number of application categories is a single patient,which basically shows a growth every year.The results mean that since 2016,the United States has continued to improve the construction of relevant regulations and promoted the development of individual patients applying for extended clinical trial drugs.
1.1.6 Charges
US pharmaceutical companies charge the subjects after approval by FDA,including management fees and production and transportation costs of related materials.One year after approval is the charging period,which can be extended,but pharmaceutical companies can also choose to provide it free of charge.
1.2 Extended clinical trial system in the European Union
According to the terminology adopted by EU’s regulations,the use of unauthorized drugs in projects involving patient groups is called compassionate medication[9].
1.2.1 Development of extended clinical trials in the European Union
In 2001,the European Medicines Agency (EMA)made it clear for the first time that investigational drugs could be used in specific situations with patients[10].In 2004,the EU’s regulation for the first time introduced the compassionate use programme(CUP) to clarify that patients can be provided with medicines that are included in the EMA centralized approval because of the principle of compassion.Besides,drugs that are explicitly included in the EMA centralized approval procedure can be provided to the patient group on the grounds of compassion[11].The European Parliament and the Council allow the use of unapproved drugs in individual patients,for example,the compassionate use for designated patients.In 2007,the EMA Committee for Medicinal Products for Human Use (CHMP) issued the “Guidelines for Compassionate Use of Medical Products”[12].Subsequently,compassionate medication guidelines for a number of specific projects were published.In some countries,doctors can act as initiators,which is a “named patient programme(NPP)”
The EU differentiates between various numbers of patients through NPP and CUP,thus constituting the EU’s extended clinical trial system.Before 2010,most countries in the European Union had issued corresponding regulatory documents,and the current system is relatively mature.
1.2.2 Scope and principles of application
Because the extended investigational drugs are not listed and their safety and efficacy cannot be guaranteed,they should not be widely used,and their applicable conditions are more stringent,including: (1)The patient has no other effective treatment methods;(2) The patient does not meet the access criteria for clinical trials of the drug;(3) The drug has a certain basic safety and efficacy data support;(4) The benefits of the drug outweigh the risk.
1.2.3 Principal responsibilities
The EU’s extended clinical trials regime includes both NPP and CUP regimes.
NPP refers to the application made by doctor for less than 50 patients according to the needs of treatment to,while CUP means pharmaceutical companies can apply to the drug regulatory authorities of various countries when clinical trials progress to phase II or phase III,and its scale is larger than NPP.The specific comparison between NPP and CUP is shown in Table 3 and Table 4.
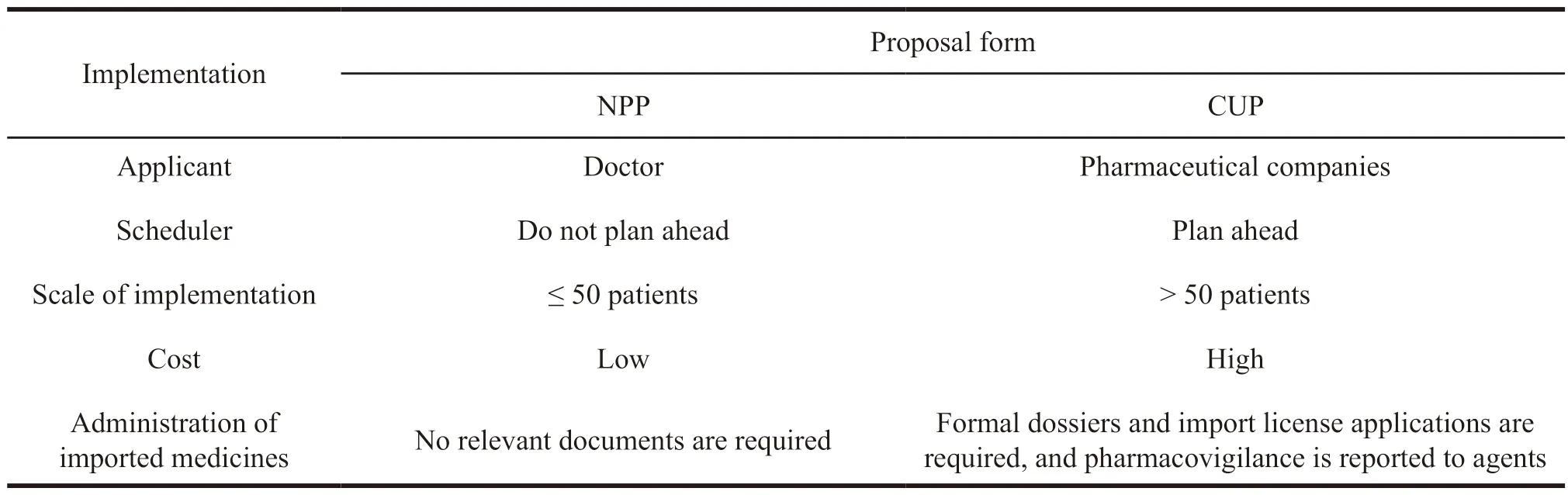
Table 3 Comparison of the implementation rules of two application methods for drugs in extended clinical trials in the European Union
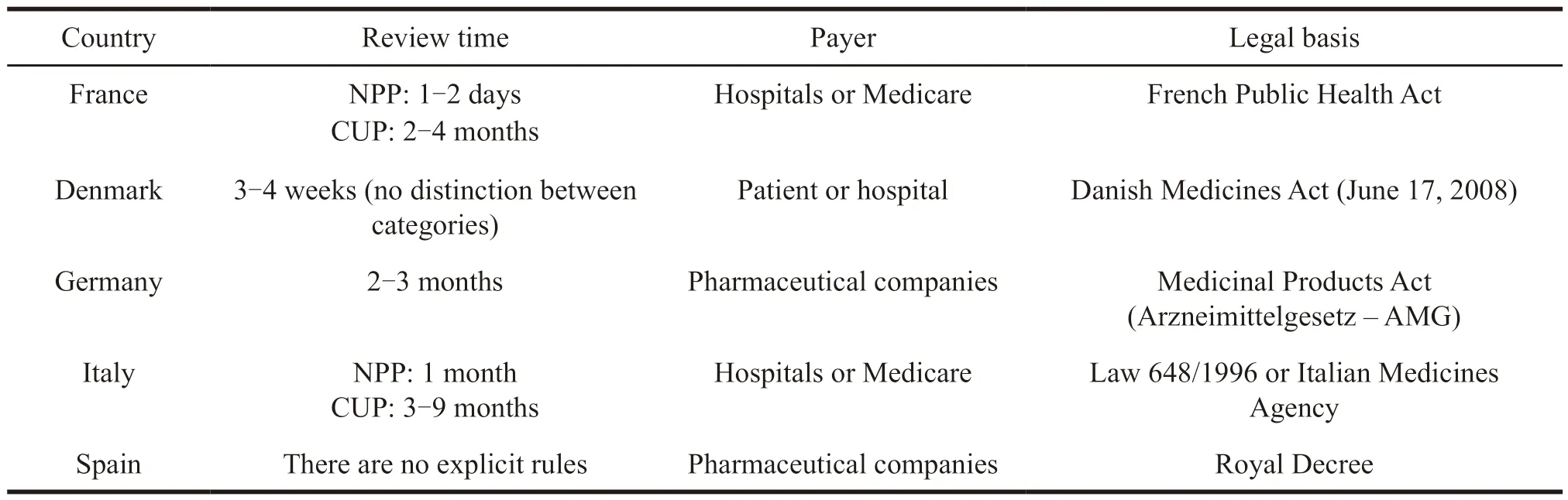
Table 4 Application methods for extended clinical trials in some EU member states
Due to the large size of CUP patients,the approval cycle is longer than that of NPP,and the management cost for pharmaceutical companies is higher.But at the same time,it is more widely used.
1.2.4 Charges
Drugs applied for in Germany,Spain and the United Kingdom are provided free of charge by pharmaceutical companies[13],while France stipulates that autorisation temporaire d’utilisation (ATU)drugs are reimbursed 100% by the French National Health Service Social Insurance,and the final price is negotiated by pharmaceutical company and the Economic Commission for Medical Products[14].
1.3 Australia’s extended clinical trials regime
Australia has two extended clinical trial regimes,the authorized prescriber scheme (APS) and the special access scheme (SAS)[15].APS is approved by the Human Research Ethics Committee or otherwise approved by the Council for Human Research to decide on the use of medicines with a history of use in Australia and no serious safety incidents within three years.And it applies to multiple patients without notice to the Therapeutic Goods Administration(TGA).SAS is an application to the TGA by a licensed pharmacist for a single patient and applies to all unapproved medicines.Since China has not yet used extended clinical trial drugs,the SAS system in Australia will be mainly introduced.
1.3.1 Australian SAS system
Under the “Therapeutic Supplies Act”,the SAS system specifically includes category A notification,category B application and category C notification.
The 3 categories of SAS channels have different applicable conditions.Category A channel is for patients with more serious diseases,which can be used without approval of the application,and the applicant only includes practicing physicians and other medical personnel representing prescribing physicians.Category C notification is for unapproved drugs with a history of use,and the list of drugs used is the drugs in the APS system,which can be notified to the TGA but can be used without approval.And category B application is applied by the medical staffto treat patient with drugs after approval by the TGA.The applicable conditions for 3 channels of SAS are detailed in Table 5 (the table applies to “√” and does not apply to “×”).In the regulations,SUSMP Schedule 8 includes government-controlled drugs.Schedule 9 is prohibited drugs,and Schedule 10 is toxic drugs.

Table 5 Comparison of the conditions applicable to 3 types of notifications/applications under the SAS system
1.3.2 Application pathways for SAS-B channels
Since SAS’s 3-channel approach varies depending on the type of patient,its flow is also different.SAS-A and SAS-C are forms of notification,while SAS-B requires filing and approval from the TGA.If SAS medication is requested,applicants can download the relevant form (different forms for the three channels) and fill it out on the TGA website.Applicants can also submit SAS application forms or notifications via the internet.And applicants can check the current processing situation (only for SAS-B classes),etc.
1.3.3 Principal responsibilities related to SAS category 3 channels
(1) Duties of the prescribing physicians.Prescribing physicians may contact TGA,patients,and sponsors to submit SAS applications for patients to qualify for unapproved medications,with responsibilities including: (1) The use of the drug is the most appropriate treatment in the current medical situation;(2) Ensure the informed consent of the patient and comply with relevant laws and regulations;(3) Monitor the medication and report to the pharmaceutical company and TGA within fifteen days[16].
(2) Duties of the TGA.TGA monitors drug use through three SAS channels,approves category B applications to search for relevant information,and encourages applicants to use medicines in the Australian Register of Therapeutic Goods (ARTG) for safety.
(3) Duties of the drug sponsors.Sponsors should provide a report every 6 months detailing the availability of the product (including quantity and frequency of supply),and adverse reactions should also be monitored and reported to TGA.
1.3.4 Payment of drug costs for SAS category 3 channels
Australia subsidizes most of the cost of medicines for most non-inpatient patients through the“drug benefit scheme”[17],but it does not subsidize extended clinical trials.Pharmaceutical companies can provide them at a low price or free of charge at their discretion.
1.4 Comparison of foreign extended clinical trials
The European Union,the United States,and Australia have all issued laws and regulations for extended clinical trials,which is shown in Table 6.
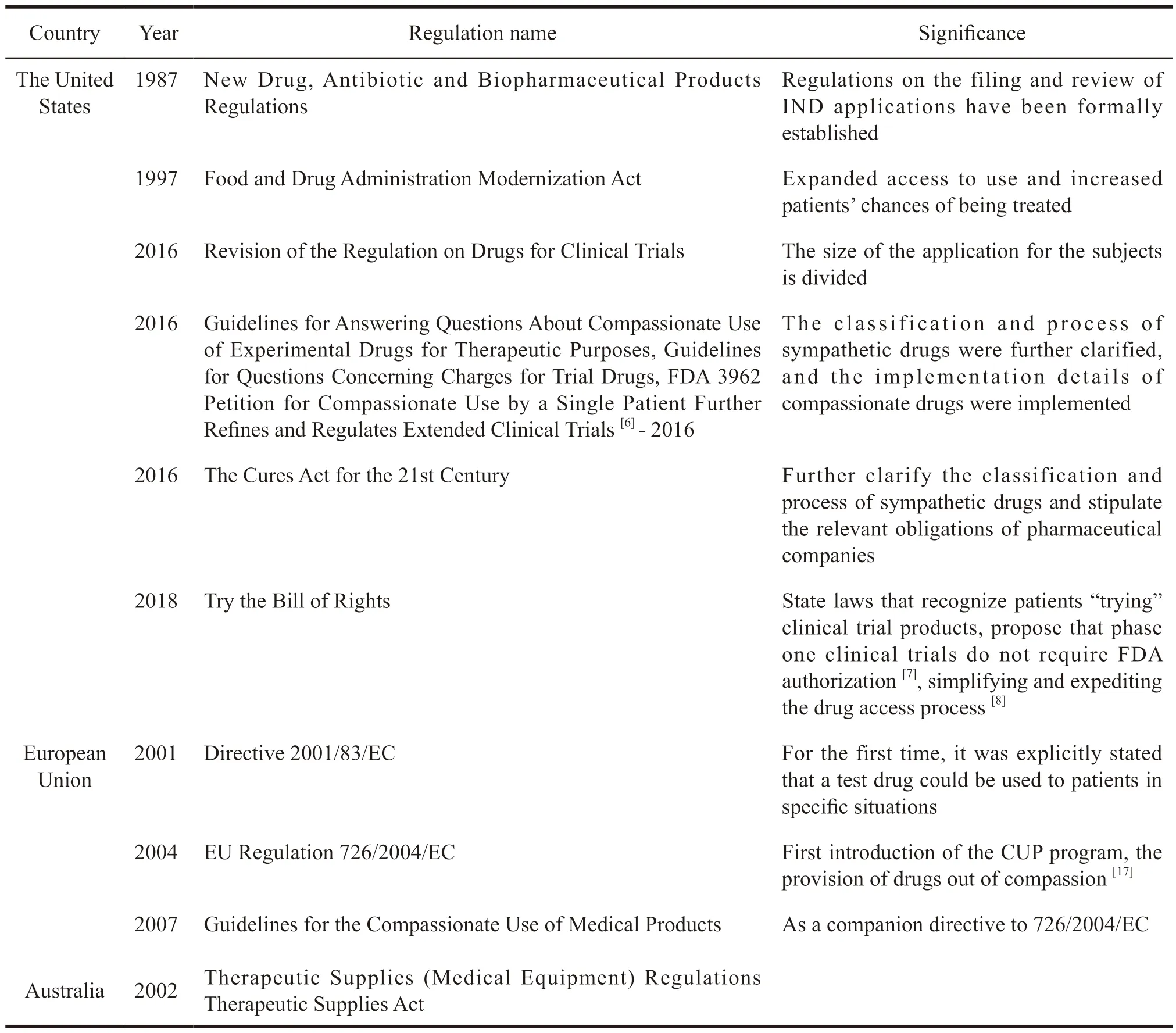
Table 6 The relevant legal system of compassionate medication in the United States,the EU and Australia
The specific implementation of the system varies according to the differences in different national regulations,as detailed in Table 7.
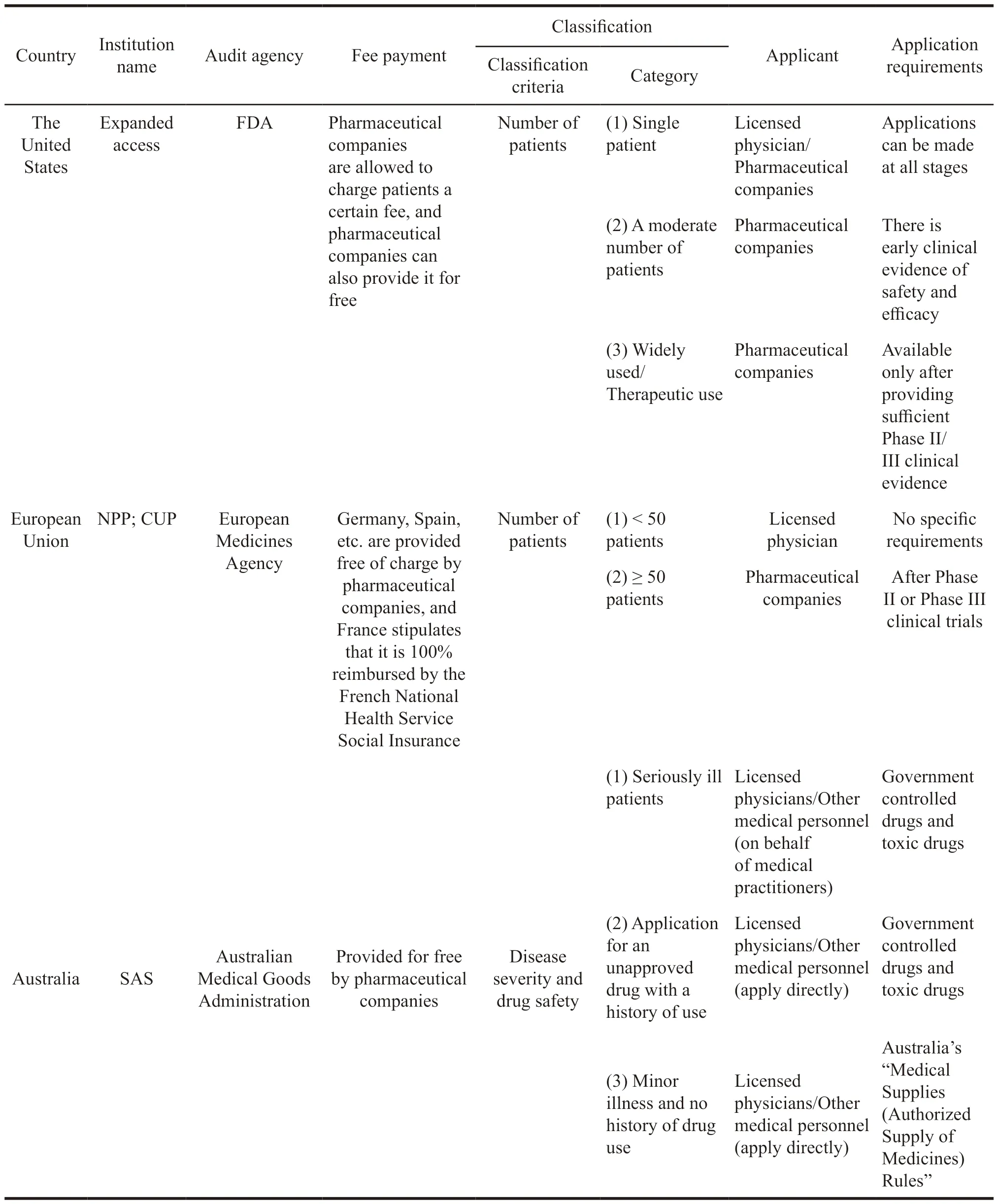
Table 7 Comparison of differences in sympathetic drug regimes in some countries
From the Table 7,it can be seen that the classification standards of extended clinical trials in different countries are not the same.The United States and the European Union are not divided based on the number of patients,and Australia is divided according to diseases and drugs.Based on this,different categories have been generated.The United States is divided into three categories: Single patient,moderate number of patients,and a large population.The European Union takes 50 patients as the dividing line,and Australia is divided into severe disease,history of use,and the other three categories.At the same time,a small number of patients in the United States and the European Union will mostly be applied by doctors,and a large number of patients apply for pharmaceutical companies.In Australia,they apply for medical personnel.The differences in classification methods also ultimately determine the differences in applicants.
2 Inspiration to China
Compared with foreign countries,due to the unclear responsibility in China,the risk is increased,and the conditions for the use of compassionate medication are more stringent.Therefore,we can appropriately increase the application channels,further improve the availability of drugs,and take different measures in a targeted manner to provide more concise methods for critically ill patients,such as through telephone,online application,approval and reply.A“fast channel” should be established to enable subjects to obtain drugs as soon as possible.At the same time,it is recommended that relevant departments build an extended clinical trial acceptance platform,and it should publicize some information so that patients can understand drug safety.For serious adverse reactions,IRB needs to conduct risk assessment in the review stage.It should determine what actions to take in the event of an emergency and assess the vigilance capability of the pharmaceutical company.

- 亚洲社会药学杂志的其它文章
- Investigation and Countermeasures of the Development of Chinese Pharmaceutical E-commerce in the B2C Model Based on PEST-SWOT Analysis
- Research on the Construction of Evaluation Model for the Development of Biopharmaceutical Park in China
- Current Situation and Prospect of the Application of Real-World Evidence in Health Care
- Study on Public Health Behavior against the Background of COVID-19 Pandemic -Based on Bandura Reciprocal Determinism
- Foreign Experience and Enlightenment of Reimbursement Management of Multi-indication Drugs
- Research on the Effect of R&D lnvestment lntensity and Sales Expense on the Performance of Biomedical Enterprises