
Zn(Ⅱ)金属有机骨架作为基质金属蛋白酶模拟物催化水解微囊藻毒素-LR
2023-12-21 01:02:10方艳芬贺玉婷牛慧斌陈春城黄应平
无机化学学报 2023年12期
方艳芬 贺玉婷 赵 丽 牛慧斌 陈春城 李 悦*, 黄应平*,
(1三峡大学材料与化工学院,宜昌 443002)
(2三峡大学三峡库区生态环境教育部工程研究中心,宜昌 443002)
(3南开大学化学学院,天津 300071)
(4中国科学院化学研究所,北京 100190)
0 Introduction
Microcystins (MCs) are a large type of cyclic heptapeptide toxins in eutrophic waters, which are commonly considered hepatotoxic and carcinogenic because of the strong inhibition of protein phosphatases and interference with cell signaling pathways. In some bodies of water that have become eutrophicated,the levels of MCs have increased to levels as high as dozens of parts per billion, which greatly exceeds the World Health Organization′s water quality standard of 1.0 parts per billion[1-4]. Advanced oxidation processes(AOPs)are capable of efficiently oxidizing MCs by producing reactive oxygen species (ROS). Nevertheless,due to the high presence of natural organic matter(NOM) in natural water bodies, AOPs are not selective to MCs because most ROS are consumed by NOM[5-7].Hydrolysis, on the other hand, is a gentler and safer way of treating pollutants,making it a preferable option over AOPs[8-11].Based on our earlier research,it was discovered that siderite, a mineral that contains iron, can break down microcystin-LR (MC-LR) peptide bonds,even in the presence of humic acid (HA), which is 10 times its amount[12]. Although siderite′s effectiveness is not very high, the results indicate that catalytic hydrolysis is a feasible approach to achieve the desired degradation of MCs when NOM is present.
Matrix metalloproteinases (MMPs), a group of proteinases dependent on Zn(Ⅱ), play a significant role in breaking down polypeptide bonds in connective tissues[13-14]. This breakdown can potentially aid in the hydrolysis of peptide bonds in MCs. Metal-organic frameworks(MOFs)hold great potential as a foundation for creating enzyme mimics or nanozymes. This is due to their remarkable versatility in composition, which enables the modification of metal ion identity and coordination environment to replicate enzymatic active sites. The functions of a series of metalloenzymes, such as tyrosinase[15], CO-dehydrogenase[16], and phosphotriesterase[17], have been performed by MOF mimics.Therefore, MOFs having functional groups can be organized in a structured manner, resulting in synergistic action, similar to MMPs. However, there are currently no artificial MMP mimics reported for MC hydrolysis,despite the widespread use of MMPs.
In this work, the physical and chemical properties of the Zn (Ⅱ)-based MOF nanomaterials (Zn-MOF-1-NSs) were meticulously analyzed and successfully used to hydroxylate MC-LR.The Zn-MOF-1-NS nanomaterial, acting as an MMP mimic, showed remarkable efficiency in catalyzing MC-LR hydroxylation with high activity and substrate specificity. Further investigation revealed that both the Zn(Ⅱ)ion and the carboxyl group of the MOF were involved in activating the peptide bond.
1 Experimental
1.1 Chemical and materials
MC-LR (Fig.S1, Supporting information) standard(1 mg solid, purity not less than 95% by HPLC) was purchased from Express Technology Co., Ltd. and stored at -25 ℃. MC-LR stock solution was prepared by adding H2O to dissolve the MC-LR standard and then stored at 4 ℃. The concentration of the MC-LR solution used inin-situATR-FTIR experiments was 250 mg·L-1and that in the degradation experiments was 2 mg·L-1. Deuterium oxide (D2O, 99.9%) was purchased from Cambridge Isotope Laboratories, Inc.Glutamic acid (D-Glu), arginine (L-Arg), alanine (DAla) and leucine (L-Leu) were obtained from Sigma-Aldrich. River humic acid standard (IR105H) was purchased from the International Humic Substances Society (IHSS). Methanol and acetonitrile were HPLC grade, and water was purified by reverse osmosis and deionization. All chemicals were obtained commercially and used as received without further purification.
1.2 Preparation of Zn-MOF-1-NS
Zn-MOF-1 precursor was synthesized by the following procedures[18]: a mixture of ZnSO4·6H2O (143.8 mg, 0.5 mmol), 3-amino-1,2,4-triazole-5-carboxylic acid (38.5 mg, 0.3 mmol), and 1,3,5-benzenetricarboxylic acid (42 mg, 0.2 mmol) was dissolved in water(10.0 mL), and placed in a 23 mL Teflon-lined stainless steel vessel.The pH of the solution was adjusted to 6.0 by triethylamine.This mixture was sealed and heated at 150 ℃for 48 h.After cooling to 25 ℃,the precipitate of the crude product was collected, washed with H2O and methanol, and dried in a vacuum at 60 ℃for 6 h. Anal. Calcd. for Zn-MOF-1 (C22H18N8O15Zn4)(%):C, 29.49; H, 2.02; N, 12.51. Found(%): C, 29.17; H,2.08;N,12.65.
The exfoliation of Zn-MOF-1 was carried out by sonicating the ethanol suspension of its precursor (8 mg in 7 mL)for 3 h.After the centrifugation at 4 000 r·min-1for 10 min to remove unexfoliated particles,Zn-MOF-1-NSs were separated from the ethanol supernatant by rotatory evaporation.
1.3 Characterization
Powder X-ray diffraction (PXRD) patterns were recorded on a Rigaku D/max-2500 diffractometer with CuKαradiation (λ=0.154 06 nm) at 40 kV and 100 mA with a scanning range of 5°-50°.Scanning electron microscopy (SEM) images were taken using a JEOL JSM-7500F scanning electron microscope at an accelerating voltage of 30 kV.Elemental analysis (C, H, and N) was performed on a Perkin-Elmer 240C analyzer.Thermogravimetric analysis (TGA) was operated with a Rigaku standard TG-DTA analyzer from ambient temperature to 700 ℃with a heating rate of 10 ℃·min-1in the air, and an empty Al2O3crucible was used as the reference. Atomic force microscopy (AFM) was carried out on a Bruker Icon probe microscope in tapping mode in the air under ambient conditions, and the delaminated MOF samples were deposited on aluminum-coated silicon cantilevers. Theζpotential of the catalyst was measured using a Malvern Zeta Sizer. N2sorption isotherms were measured with a Micromeritics ASAP 2020 analyzer at 77 K. X-ray photoelectron spectroscopy (XPS) was conducted on an AXIS Supra spectrometer equipped with 300 W AlKαradiation.The hydrocarbon C1sline at 284.8 eV from adventitious carbon was used for energy referencing.A Nicolet iS50 Fourier transform infrared spectrometer collected infrared spectra(FTIR).
1.4 Hydrolysis experiments
Reactions were carried out in a 10 mL vial placed in a dark box with magnetic stirring at (25±0.1) ℃. In each experiment, 3 mL aqueous solution of MC-LR with the initial concentration of 2 mg·L-1was placed in the vial, and 5 mg catalyst was dispersed to it. The pH of the suspension was not adjusted. At specified reaction times, 400 μL samples of the reaction mixture were collected, centrifuged to remove the catalyst, and analyzed using HPLC.
The substrate specificity of Zn-MOF-1-NS toward MC-LR in the presence of NOM was tested with the addition of HA with 1, 2, 10, and 20 mg·L-1. Other reaction and analytical conditions were unchanged.
1.5 Analytical methods
MC-LR. A Waters 2690 HPLC determined the concentration of MC-LR with a PDA detector and a Kromasil C18 column (4.6 mm×250 mm, 10 μm particle size). The mobile phase was water and methanol(60∶40,V/V) containing 0.05% trifluoroacetic acid(TFA). The flow rate, injection volume, and column temperature were 0.8 mL·min-1, 20 μL, and 35 ℃,respectively. MC-LR was quantified by its absorption at 238 nm. The retention time of MC-LR was 8.3 min.The quantification range was from 0 to 10 μmol·L-1for MC-LR with the correlation coefficient(R2)for the standard of 0.998(Fig.S2a).
Amino acids. Before measurement, 0.6 mL of 4∶1(V/V) water-ethanol mixture was added to wash the adsorbed amino acids from the MOF catalyst (Fig.S3)[19].The details about desorption experiments are provided in the Supporting information. The hydrolysis products(D-Glu,L-Arg,D-Ala, andL-Leu) of MC-LR were determined by a Waters 2690 HPLC with an FLR detector and a Nova-PakTMC18 column (3.9 mm×150 mm, 4 μm particle size). The retention times ofD-Glu,L-Arg,D-Ala, andL-Leu were 8.3, 14.3, 19.8, 21.3,and 31.8 min, respectively. The hydrolysis products were quantified using fluorescent detection withλex=330 nm andλem=445 nm. These compounds were identified and quantified by the external standard curves of the peak area. The quantification ranges for four amino acids were from 0.5 to 20 μmol·L-1with theR2for the standards above 0.99(Fig.S2b).
1.6 In-situ ATR-FTIR analysis
In-situinfrared spectroscopy was used to monitor the surface interaction between Zn-MOF-1-NS film and MC-LR in different solvents (H2O,D2O,and C6H6).The ATR cell was placed in the internal compartment of a Nicolet iS50 FTIR spectrometer (Thermo, USA)equipped with an MCT-A detector.Thin Zn-MOF-1-NS film was prepared by drying Zn-MOF-1-NS suspension(0.4 mL, 10 g·L-1) on a ZnSe crystal strip (80 mm×10 mm×4 mm). After adding 0.8 mL of solvent to the flow cell, ATR-FTIR spectra were collected continuously for 2 or 4 h until the establishment of the sorption equilibrium between solvent and Zn-MOF-1-NS film. After MC-LR (100 μL, 250 mg·L-1) was added, the timeresolved ATR-FTIR spectra were recorded at the interval of 0.283 s.
1.7 Theoretical calculation
The geometry optimization, frequency calculation,and natural bond orbital (NBO) analysis were all performed at the M06/def2-TZVP level[20-21]. To make the calculation affordable, besides the central Zn2+ion,only the ligands coordinating directly with it were considered in treating Zn3 and Zn4. For Zn2, because of the possibility of H2O/reactant forming an H bond with nearby carboxyl groups, two additional btc3-ligands were taken into the model. The initial configurations of Zn2+and ligands were picked up from the crystalline structure of Zn-MOF-1. Since the peripheral carboxyl groups of btc3-ligands are far from the reaction center of Zn2+, they were removed with H atoms supplemented to the vacancies. During optimization, the C atoms in the benzene rings and the N atoms coordinating to peripheral Zn2+ions (not included in the model) were fixed to replicate the bulk behavior, while the other atoms were relaxed (Fig.1). An implicit solvent model of SMD (solvation model density) modulated the solvent effects.
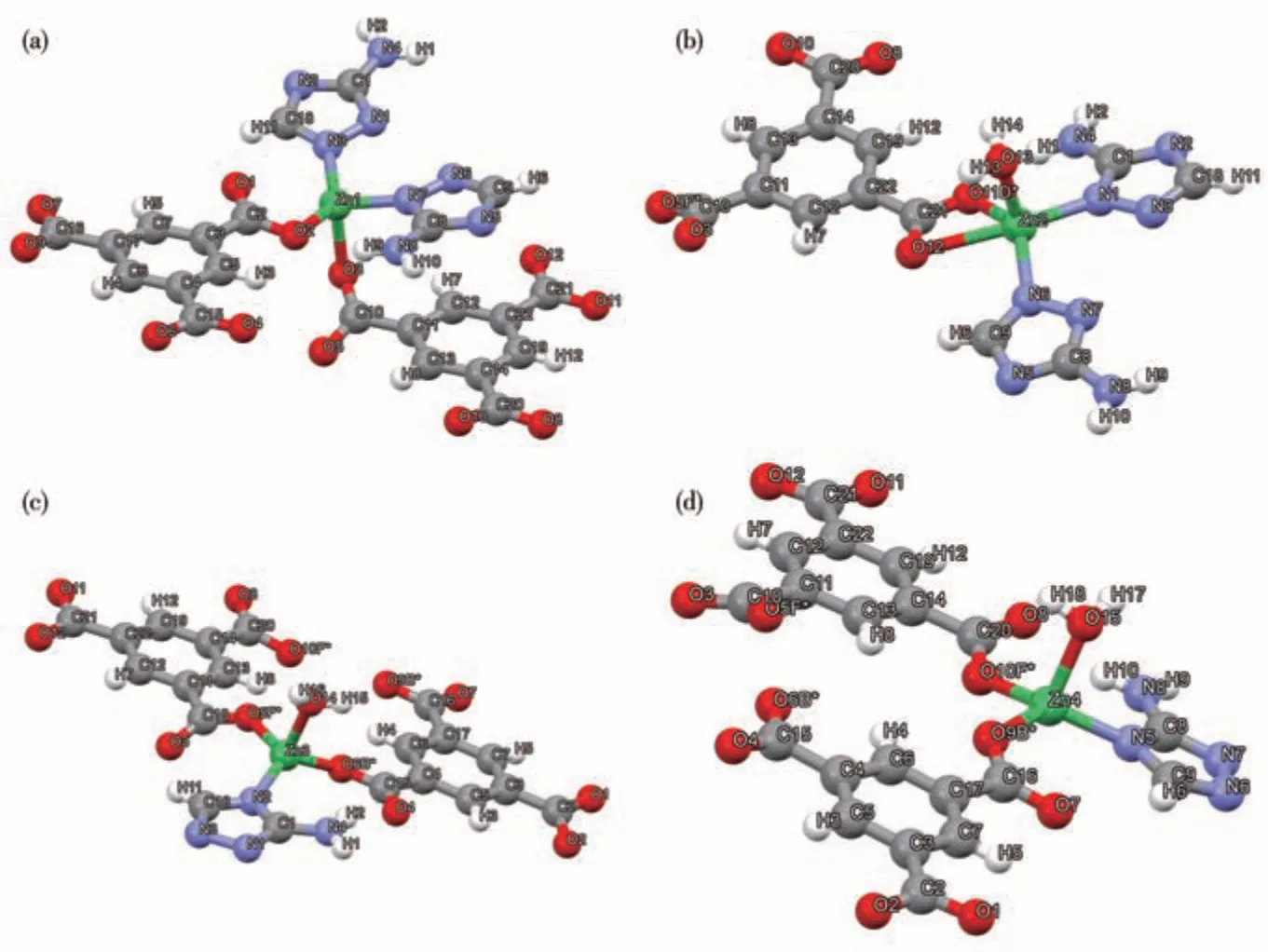
Fig.1 Coordination environments of(a)Zn1,(b)Zn2,(c)Zn3,and(d)Zn4 in Zn-MOF-1-NS
2 Results and discussion
2.1 Selection of MOF catalyst
Due to the large molecular size of MC-LR (2.4 nm), it is difficult to design channels large enough to accommodate MC-LR molecules without affecting the stability of the MOF. The results reported by Tissot et al.[22]and our previous works[23]have shown the inefficiency of internal active sites towards large-molecular reactants. Therefore, we chose 2D nanosheets, which expose the maximum quantity of active sites on the external surface, as the morphology for the MOF catalyst.Combining with the structure and catalytic mechanism of MMP,the selection criteria of the MOF catalyst were: (1) the backbone is constructed by the connection among Zn2+ions and five-membered N-heterocyclic bridging ligands, and carboxyl groups are contained for the activation of peptide bond;(2)in addition to heterocyclic N atoms, Zn2+ion is coordinated with at least one solvent molecule, which would be easily replaced by substrate and does not destroy the integrity of the MOF; (3) the framework is 2D connected, which could be delaminated to nanosheets. After the screening of the 424 MOFs containing Zn2+and N-pentacyclic ligand containing MOFs summarized in the CCDC database, [Zn4(atz)2(btc)2(H2O)3]n(Zn-MOF-1), a MOF assembled by the ligands of 3-amino-1,2,4-triazolate(atz-) and 1,3,5-benzenetricarboxylate (btc3-), was chosen as a potential catalyst for the following investigation (Fig.2)[18]. This MOF contains four crystallographically independent Zn(Ⅱ)centers (Fig.1). The benzene ring and triazole ring projections on the ab plane are staggered, but there isn′t any effective overlap. Therefore,theπ-πstacking doesn′t play a significant role in the crystal stability of this MOF.All of these Zn(Ⅱ)centers are surrounded by triazole N atoms and carboxyl O atoms, and three of them (Zn2, Zn3, and Zn4) are coordinated by one H2O molecule. The interconnection among Zn2+ions and two kinds of ligands form a double-layer structure, which expands into the supramolecular framework of the crystal only by the weak connection of H bonds. The 2D connected structure of Zn-MOF-1 was drawn with Mercury 4.2.0.

Fig.2 Two-dimensional connected structure of Zn-MOF-1
2.2 Preparation and characterization of MOF nanosheets
Zn-MOF-1 was synthesized generally according to the reported procedure with minor modifications to minimize the particle size[18]. PXRD was applied to characterize the obtained sample (Fig. 3a). All the recorded diffraction peaks could be readily indexed to the simulated pattern, showing that our sample presents the same framework structure and has good phase purity. This backbone was further supported by the results of elemental analysis, thermogravimetric (Fig.S4),and FTIR analyses(Fig.S5).Because of its 2D connected framework, the sample of this MOF presented a layer-stacking morphology under SEM observation(Fig.3b). In consistent with its nonporous structure,Zn-MOF-1 presented a small BET surface area of 8 m2·g-1(Fig.S6a and Table S1). This result implied that Zn-MOF-1 has limited contact with the reactants,so its delamination is necessary.
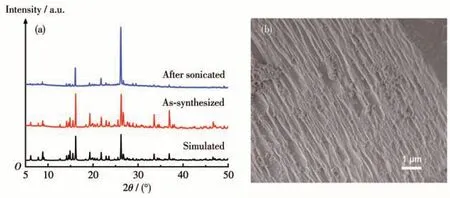
Fig.3 (a)XRD patterns and(b)SEM image of Zn-MOF-1
The Zn-MOF-1 precursor was exfoliated simply by the sonication in ethanol (the product is denoted as Zn-MOF-1-NS).After the removal of sediments,a transparent supernatant was obtained. The exfoliation process was evidenced by an apparent Tyndall effect under the irradiation of a laser beam (Fig.4a).The morphology of the exfoliated sample was examined by AFM.As shown in Fig.4b and 4c,this sample was composed of irregularly shaped nanosheets with several hundred nanometers lateral dimensions.The thicknesses of these nanosheets were generally about 4 nm,indicating that each nanosheet comprises only six two-layer structures of Zn-MOF-1. Zn-MOF-1-NS presented a negativeζpotential of -9.2 mV at pH=7.4 (Fig.S7),which ensured its good stability in the aqueous suspension[24].
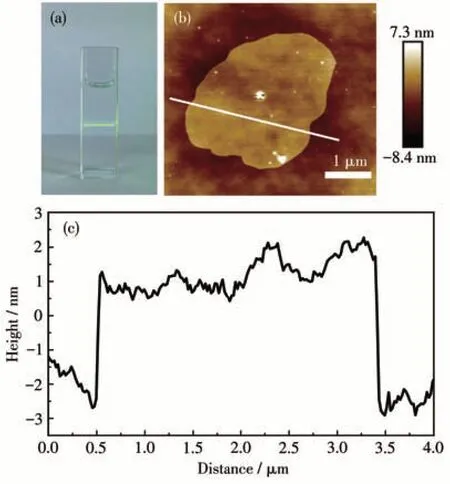
Fig.4 (a)Photograph of the Tyndall effect of the ethanol suspension of Zn-MOF-1-NS;(b)AFM image of Zn-MOF-1-NS;(c)Height profile of the corresponding section of figure b
2.3 Catalytic degradation of MC-LR
With 5 mg of Zn-MOF-1-NS, 82.6% of MC-LR was degraded in 7.5 h(Fig.5a).MC-LR decay generally followed first-order kinetics withR2=0.94 (Fig. 5b).Within 7.5 h, only 20.9% of MC-LR was degraded by siderite, indicating that the activity of Zn-MOF-1-NS was much higher than those of siderite that showed the highest activity reported for the hydrolysis of MC-LR[12].To further examine the reuse potential of Zn-MOF-1-NS, cyclic degradation experiments were carried out(Fig.5d).It was observed that Zn-MOF-1-NS continued to display high catalytic activity in five successive cycles. However, due to the high dispersity of nanosheets,part of the catalyst was lost during the recycling process, which induced the slowdown of the reaction(by 45% in five cycles).Furthermore,as evidenced by the PXRD pattern (Fig.S8),the Zn-MOF-1-NS catalyst maintained good stability during the reaction.
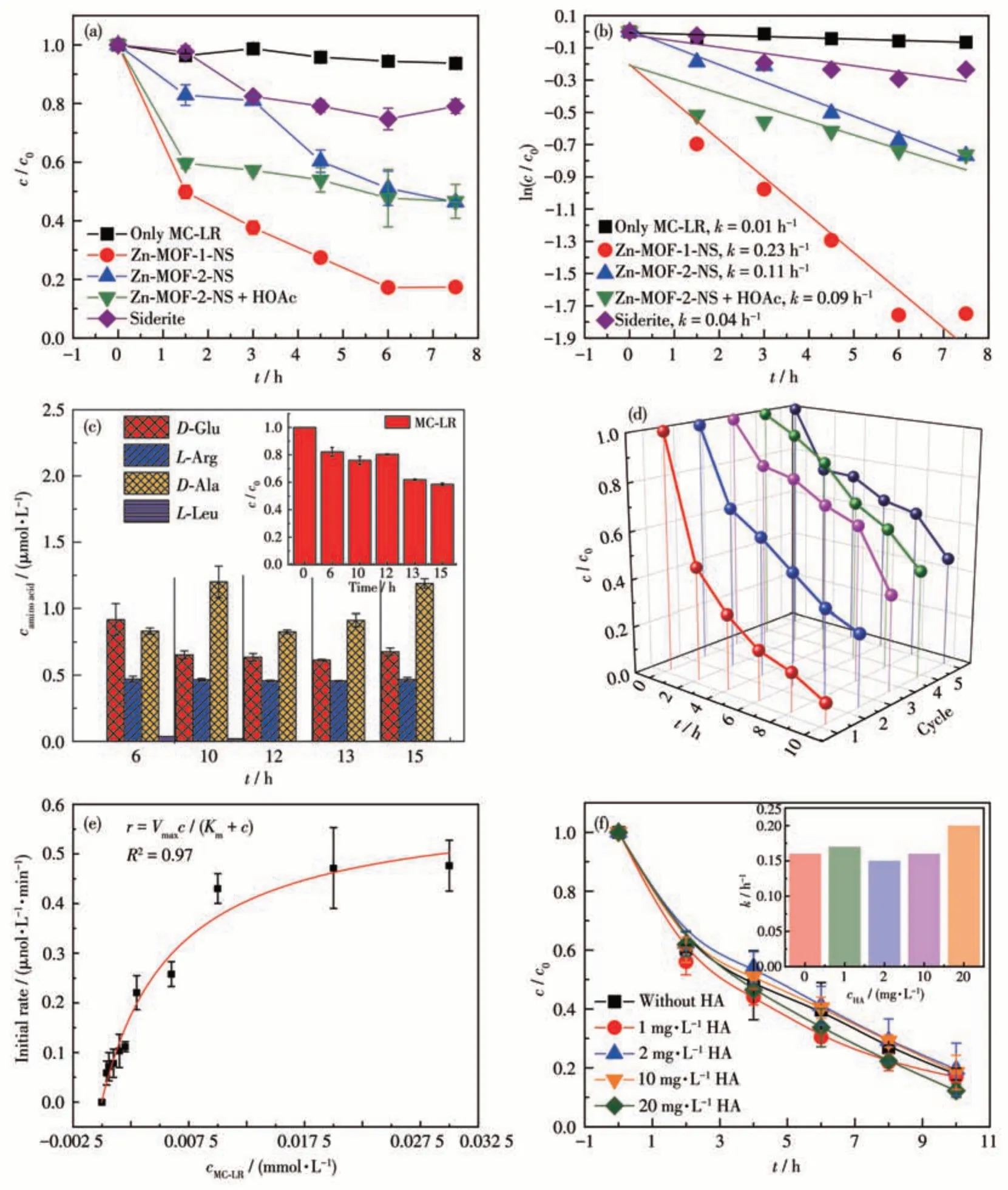
Fig.5 (a)Degradation curves and(b)reaction kinetics of MC-LR using various catalysts;(c)Hydrolysis yield production of amino acids using Zn-MOF-1-NS(Inset:MC-LR degradation curve);(d)Recycling of Zn-MOF-1-NS;(e)Michaelis-Menten kinetic curve of the initial reaction rate against MC-LR concentration;(f)Degradation curves of MC-LR(2 mg·L-1)at different concentrations of HA in Zn-MOF-1-NS(Inset:effect of HA concentration on the reaction rate constant of MC-LR hydrolysis)
To illustrate the reaction mechanism, another 2D Zn(Ⅱ)based MOF,[Zn7(OH)2(Hbta)6(H2O)6]n(H3bta=N,Nbis-(1(2)H-tetrazol-5-yl)-amine) (named Zn-MOF-2)was also tested for comparison[25]. The characterization of Zn-MOF-2 is also shown in Fig.S9-S11. Under Zn-MOF-2-NS catalysis, the degradation efficiency of MC-LR in 7.5 h could only reach 54.0%,obviously lower than the result of Zn-MOF-1-NS. The Zn2+density could not explain the superior activity of Zn-MOF-1-NS because the surficial Zn2+density of Zn-MOF-1-NS(1.67×10-5nm-2)was even lower than that of Zn-MOF-2-NS (2.31×10-5nm-2). Considering that the carboxyl group was only contained in Zn-MOF-1-NS, we speculated that this group participates in the hydrolysis of MC-LR. However, the addition of acetic acid to the Zn-MOF-2-NS catalytic system could not remarkably accelerate MC - LR consumption. This phenomenon indicated that the relative positions of Zn2+and the carboxyl group are crucial.
The intermediates produced during the reaction were tested with an initial concentration of MC-LR of 10 mg·L-1.D-Glu,L-Arg,D-Ala, andL-Leu were detected during the decomposition of MC-LR (Fig.5c).At 10 h, the concentrations were 0.653, 0.466, 1.20 and 0.0183 μmol·L-1, respectively. The quantity ofD-Ala could account for 50.0% of MC-LR consumption, confirming that the hydrolysis of peptide bonds was the main pathway for MC-LR degradation.
A steady-state kinetic assay was performed by varying the initial concentration of MC-LR. The relationship between the initial reaction rate and substrate concentration could be well described by the Michaelis-Menten equation withR2=0.97, showing the enzyme mimic feature of Zn-MOF-1-NS (Fig.5e). The fitting gave the Michaelis constant (Km) of 6.2 μmol·L-1and the maximum reaction rate (Vmax) of 0.61 μmol·L-1·min-1. SinceKmis the substrate concentration at which half the enzymes are bound and its values for MMPs range from tenths to tens of millimoles[26-29], this result indicated that the affinity of Zn-MOF-1-NS for MC-LR is comparable with those of MMPs towards proteins.
Various NOMs are contained in environmental water samples and could potentially inhibit MC-LR degradation. HA was added to the reaction system to test the ability to resist NOM interference of Zn-MOF-1-NS. Even when the HA concentration was increased to 20 mg·L-1, the rate change of MC-LR (2 mg·L-1) was still negligible (Fig.5f), showing that Zn-MOF-1-NS can specifically degrade MC-LR in the presence of NOMs. In contrast, with Zn-MOF-2-NS as a catalyst,the same concentration of HA led to a significant slowdown of the reaction (the percentage of degradation decreased from 46.7% to 29.5%, Fig.S12). Based on the structure of Zn-MOF-1-NS, this catalyst was expected to catalyze the hydroxylation of the peptide bond by the Lewis acidity of Zn2+ions. However, our previous research illustrated that a single Lewis acid site of mineral catalysts did not display specificity for MC-LR[12].Thus, the phenomenon in the Zn-MOF-1-NS catalytic system indicates the existence of multiple binding processes in the reaction.
2.4 Hydrolysis of MC-LR on Zn-MOF-1-NS surface
In the ATR-FTIR spectrum of Zn-MOF-1-NS(Fig.S13a),the wide band at 3 260 cm-1includes O—H and N—H stretching vibrations[30]. The 1 615 and 1 431 cm-1peaks belong to the C=O and C—O stretching vibrations of COO-that coordinate with Zn2+, respectively[12,24]. The vibration at 1 554 cm-1is attributed to N—H[31]. The band at 1 369 cm-1belongs to the C—C bonds of the benzene ring[32]. The absorptions at 1 211 and 1 062 cm-1correspond to the C—N bonds on and in the triazole rings, respectively[33]. The stretching vibration of Zn—O appeared at 734 cm-1[34]. The spectrum of MC-LR is given in Fig.S13b. In this spectrum,the absorption bands at 3 260 and 1 515 cm-1refer to the stretching and bending vibrations of N—H[12]. The bands at 2 925 and 2 851 cm-1are assigned to the stretching vibrations of —CH2—[35]. The C=O stretching vibration of COOH was centered at 1 731 cm-1[36].The peak at 1 642 cm-1(Ⅰband) and 1 450 cm-1(Ⅱband)was ascribed to the amide C=O stretching vibration[37]. The peak at 1 642 cm-1is ascribed to the amide C=O stretching vibration (Ⅰband). The vibrations of C—H in-plane bending, C—O—C stretching, and C—N stretching correspond to the peaks at 1 409, 1 178,and 1 082 cm-1,respectively[25].
Fig.6a and 6b show thein-situATR-FTIR spectra of the Zn-MOF-1-NS/H2O equilibrium process, which were recorded at intervals after MC-LR addition (100 μL, 250 mg·L-1). The positive bands at 3 501, 3 409,1 648, 1 537, 1 487, 1 401, and 1 087 cm-1showed that MC-LR is adsorbed on the surface of Zn-MOF-1-NS.A redshift of the peak of the amide C=O of MC-LR from 1 648 to 1 637 cm-1was observed,which could be explained by its coordination with the Zn2+of Zn-MOF-1-NS. Because Zn2+works as a Lewis acid site, such coordination would reduce the π electron density of the amide group and thus weaken the C=O bond[38-39]. In addition,the negative peak at 1 615 cm-1indicated that the—COOH of Zn-MOF-1-NS is involved in the interaction with MC-LR in addition to Zn2+. Meanwhile, the new band at 3 183 cm-1illustrates the formation of a hydrogen bond between the amide N—H of MC-LR and the carboxyl oxygen of Zn-MOF-1-NS[27]. With the proceeding of the reaction, two new peaks of —COOH positively occurred at 1 601 and 1 664 cm-1[40-41], while two negative bands appeared at 1 216 and 1 065 cm-1,which are assigned to the cleavage of the amide C—N bond of MC-LR. These changes indicated that the peptide bond of MC-LR is broken into—COOH and N—H under the catalysis of Zn-MOF-1-NS.
To verify the hydrolysis mechanism,analogous experiments in D2O were performed.In the spectra of Zn-MOF-1-NS, the bands of O—H at 3 297 and 1 644 cm-1were red-shifted to 2 470 and 1 201cm-1for the O—D bond (Fig.S14), which conformed to the Hooke′s law[42]. Upon the addition of MC-LR, the same trend as that in the Zn-MOF-1-NS/H2O system was observed(Fig.6c). A crest appeared at 1 664 cm-1, showing the formation of —COOH by the cleavage of amide C—N[43-44]. The peak of N—H bending vibration at 1 330 cm-1and the valley of the break of the C—N bond at 1 047 cm-1confirm the cleavage of the peptide bond of MC-LR (Fig.6d). The redshifts of the —COO-peak of Zn-MOF-1-NS from 1 431 cm-1(in the pure Zn-MOF-1-NS/D2O system, Fig.S13a) to 1 450 cm-1and the N—H peak of MC-LR from 1 554 to 1 330 cm-1confirm the formation of hydrogen bond between them. Together with the results of the H2O system,these changes manifest that both Zn2+and—COO-of Zn-MOF-1-NS participate in the interaction with MC-LR.The former acts as a Lewis acid site to coordinate with amide C=O and the latter binds to N—H through a hydrogen bond.
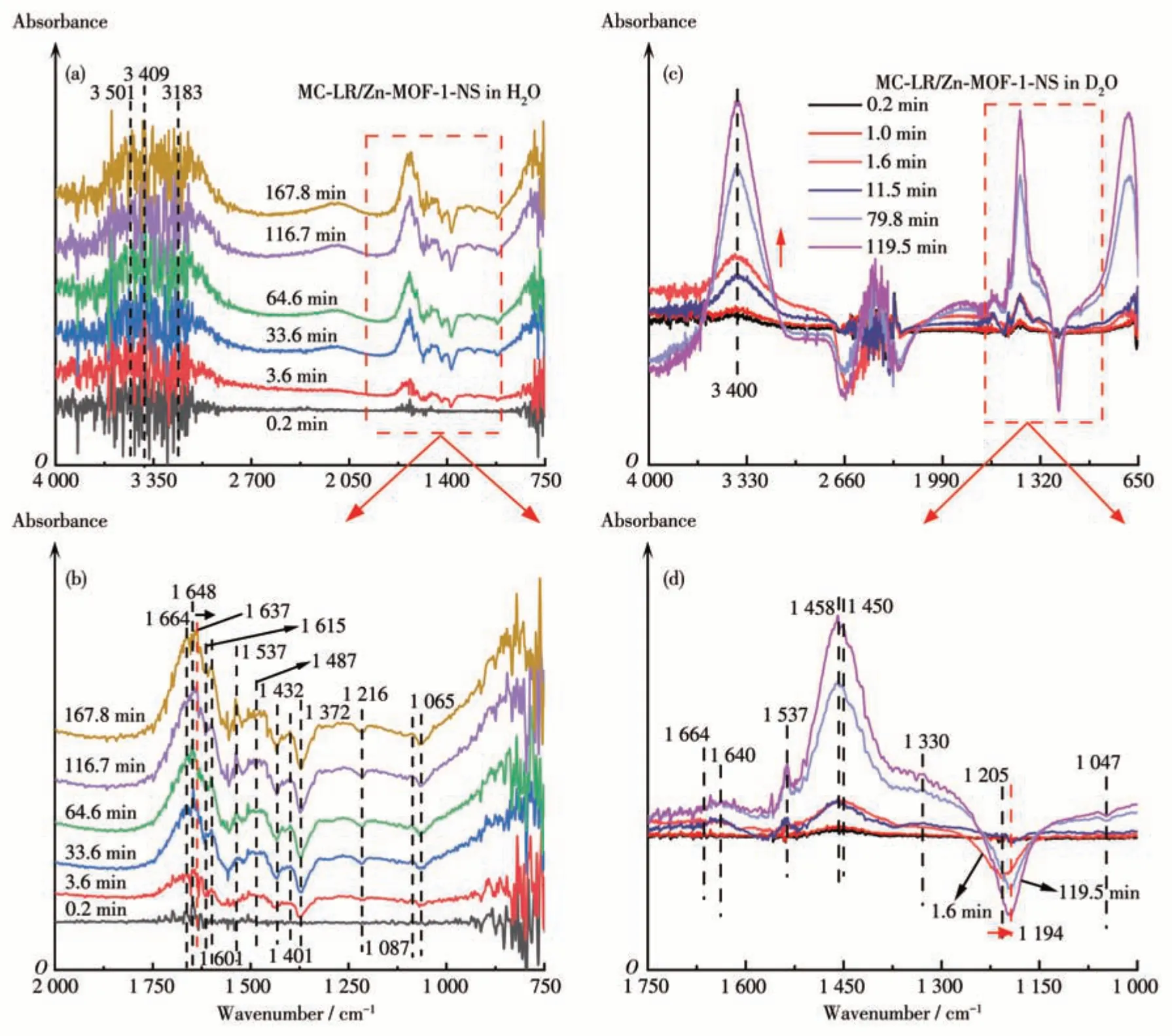
Fig.6 In-situ ATR-FTIR spectra of(a,b)MC-LR(100 μL,250 mg·L-1)in H2O system and(c,d)MC-LR(100 μL,250 mg·L-1)in D2O system
In addition, the infrared spectra of Zn-MOF-1-NS separately immersed in H2O and D2O were recorded for comparison (Fig.S15). Relative to the system of H2O,two new peaks were observed at 2 488 and 1 246 cm-1in D2O, indicating that D2O replaced the coordinating H2O molecule. Therefore, it is necessary to study the fate of MC-LR after binding with Zn2+sites in the D2O system.After the addition of MC-LR to the D2O system,thein-situATR-FTIR spectra presented a valley at 1 205 cm-1(O—D, for D2O desorption) and the crests at 3 400 cm-1(N—H of adsorbed MC-LR) and 1 450 cm-1(C=O of amide Ⅱ) from 0.2 to 119.5 min(Fig.6d), indicating that the D2O coordinating to Zn2+is replaced by MC-LR.
Similar phenomena were also observed in the benzene system. Zn-MOF-1-NS equilibrated at C6H6for 2 h (Fig.7a). The loss of the coordinating H2O molecule in Zn-MOF-1-NS gave negative bands of H—O stretching at 3 386 and 1 632 cm-1(Fig.7b).At the same time,it was observed that the Zn—O band presented a blue shift from 774 to 804 cm-1. This change confirms the replacement of the H2O molecule by MC-LR on the Zn2+site.
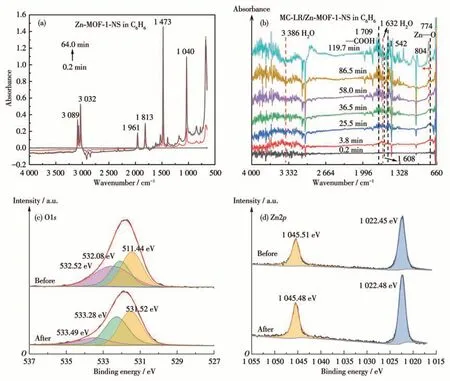
Fig.7 In-situ ATR-FTIR spectra of(a)Zn-MOF-1-NS equilibrated for 2 h and(b)MC-LR(100 μL,250 mg·L-1)/Zn-MOF-1-NS in C6H6 system(Solvent-Zn-MOF-1-NS spectrum as background);XPS spectra of Zn-MOF-1-NS before and after the reaction with MC-LR for 10 h:(c)O1s;(d)Zn2p
We further used XPS spectroscopy to investigate the interaction between MC-LR and Zn-MOF-1-NS.For pristine Zn-MOF-1-NS, three peaks of O1swere recorded at 531.44, 532.08, and 532.52 eV, corresponding to Zn—O[45], O—H[46], and H2O groups[47],respectively (Fig.7c). After the addition of MC-LR, the peak of H2O shifted to 533.49 eV and its area decreased(from 36.1% to 16.0%), verifying the replacement of H2O molecule by MC-LR. Because of the formation of hydrogen bonds with MC-LR,the peak of the O atom of Zn—O presented a blue shift to 532.28 eV. Considering that Zn2+works as the Lewis acid site for MC-LR,we also examined the Zn2pspectrum(Fig.7d).The pristine MOF gave two peaks at 1 022.4 and 1 045.5 eV,which could be assigned to the Zn2p3/2and Zn2p1/2spinorbit components of O/N coordinated Zn atom, respectively[48-49]. Since replacing the coordinating aqueous O atom with the amide O atom of MC-LR does not change the first coordination sphere of Zn2+, the addition of MC-LR did not significantly alter the positions of these two peaks.
The experimental observations indicate: (1) the much higher activity of Zn-MOF-1-NS than that of Zn-MOF-2-NS proved the participation of the carboxyl group; (2) the substrate specificity of Zn-MOF-1-NS towards MC-LR illustrates the formation of multiple bonds between them; (3) FTIR analysis shows that the amide O atom coordinates to Zn2+ion, and N—H is hydrogen bounded; (4)in-situATR-FTIR and XPS spectra indicate the coordination of amide O atom of MC-LR to Zn2+with the replacement of H2O molecule.So, we speculate that the catalytic mechanism of Zn-MOF-1-NS is similar to that of MMP and involves the participation of both Zn2+ions and carboxyl groups.Thus, we resorted to the theoretical calculation to give an atomic insight into the activation mechanism of MC-LR by Zn-MOF-1-NS. Since the reaction occurred mainly around the peptide bond, N-methylacetamide was taken as a simplified model for MC-LR. As introduced above, among the four crystallographically independent Zn(Ⅱ)centers in Zn-MOF-1-NS,three of them,Zn2, Zn3, and Zn4, are coordinated by H2O molecules and can provide coordination sites for amide O atom.Therefore, we first calculated the Gibbs free energy change (ΔG) values for the substitution of H2O molecule byN-methylacetamide on these metal sites, and the results are listed in Table 1. Due to the similar coordination environments of Zn3 and Zn4, they gave close ΔGvalues of -10.4 and -11.6 kJ·mol-1, respectively.The negative values of these free energy changes show that the coordination of amide to these Zn sites is thermodynamically feasible. The scrutinization of the geometries of these complexes shows that besides the coordination of the amide O atom to Zn2+(Zn—O:0.208 nm for Zn3 and 0.204 nm for Zn4), the N—H also forms a strong hydrogen bond with the carboxyl O atom in the ligand (O…H: 0.198 and 0.202 nm for Zn3 and Zn4, respectively, and ∠N—H…O=163.4° and 157.9°,Fig.S16).
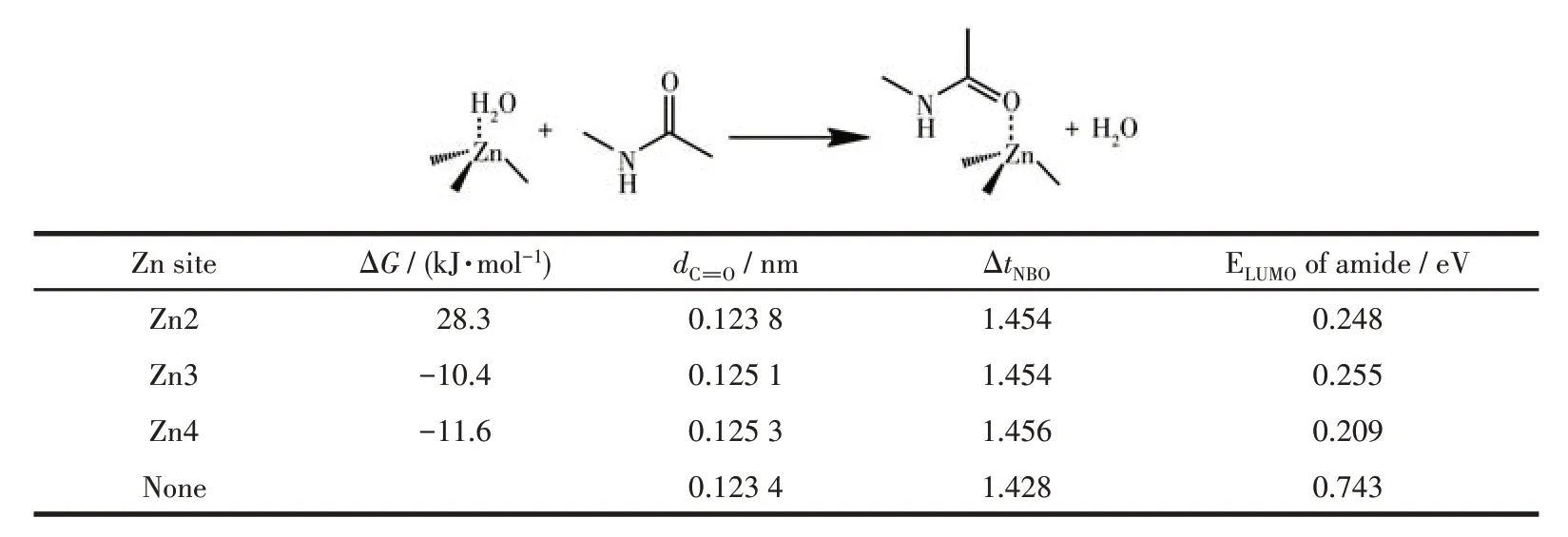
Table 1 Results of the calculation about amide adsorption to Zn sites of Zn-MOF-1-NS
In contrast, Zn2 provided a positive ΔGfor the binding with amide (28.3 kJ·mol-1), which may result from the position of its coordinating H2O molecule.Unlike the coordinating H2O molecules of Zn3 and Zn4, which point to the outside of the double-layer structure, the coordinating H2O molecule of Zn2 is located on the opposite layer (Fig.1 and 2). This position makes the substituting amide group crowd with nearly btc3-ligands and thus weakens the formed Zn—O bond (0.226 nm). The steric hindrance also hinders the H bond formation with the carboxyl group coordinating directly with Zn2+. The amide N—H can only form a weak H bond with a carboxyl group in the second coordination sphere with longer O…H distance(0.214 nm) and narrower ∠N—H…O (141.8°). Therefore, the main active sites in Zn-MOF-1-NS are the Zn3 and Zn4 on the surface of the nanosheet, and their carboxyl ligands contribute to the binding with the amide.
Since the first step of amide hydroxylation is the nucleophilic addition of H2O to the amide C atom, the bond order and polarity of C=O[50]and the LUMO level of the amide group[51]would be parameters to predict the reactivity of the substrate. It is observed that the C=O bonds in three amide-Zn complexes are all longer than those in free N-methylacetamide, and their total NBO polarities (Δt, the difference in the NBO charge between C and O atoms)also show a remarkable increase. The LUMO level of theN-methylacetamide presents aca. 0.5 eV decrease upon the coordination with Zn sites.These results prove that,after the adsorption to the catalyst,the amide C=O bond is weakened,its C atom becomes more positive-charged and electrophilic, and the antibonding orbital of this group becomes more feasible to accommodate the lone pair electrons of nucleophilic agent (H2O), and thus the reactivity is enhanced.
3 Conclusions
In summary, simulating the structures of MMPs, a Zn (Ⅱ)- based MOF with nanosheet morphology was developed to catalyze MC-LR hydrolysis. This catalyst can rapidly remove MC-LR even in the presence of a high concentration of HA. The comparison with Zn-MOF-2-NS showed that the carboxyl groups in Zn-MOF-1-NS contribute much to the superior activity and substrate specificity.In-situATR-FTIR,XPS,and theoretical calculation proved that multiple binding processes exist between the MC-LR and Zn-MOF-1-NS and contribute to the peptide bond activation. This work demonstrates the potential of mimic enzyme catalysis in environmental treatment and that MOF is an ideal platform to simulate the multi-group synergism in enzymes.
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
党员生活·下(2022年1期)2022-04-23 21:23:01
当代音乐(2021年12期)2021-12-15 09:55:43
学生天地(2020年31期)2020-06-01 02:32:24
四川地质学报(2020年2期)2020-05-31 06:53:00
中华奇石(2016年9期)2016-12-15 14:20:46
山西农经(2016年3期)2016-02-28 14:23:53
西北园艺(果树)(2015年1期)2015-02-21 16:44:50
癌变·畸变·突变(2014年2期)2014-03-01 04:39:43
华东理工大学学报(自然科学版)(2014年3期)2014-02-27 13:48:56
华东理工大学学报(自然科学版)(2014年3期)2014-02-27 13:48:56
