
二硫化钼在电催化产氢中的应用研究进展
2023-12-18 07:24郑雍智巩鑫宇周文元王金淑
材料工程 2023年12期
郑雍智,巩鑫宇,周文元,胡 鹏,王金淑
(北京工业大学 材料与制造学部,北京 100124)
随着人类对能源需求不断增加以及化石能源所带来的环境问题持续加剧,发展绿色能源技术迫在眉睫[1-3]。氢能作为一种绿色清洁能源,具有热值高、可再生、获取途径广泛等优点,在可再生能源中占有极其重要的位置,是未来构建以清洁能源为主的多元能源供给系统的重要载体[4]。然而,不同于煤炭、石油等在自然界中天然存在的能源,氢气的制取需要依赖于其他资源,因此,开发和利用氢能技术已成为未来能源技术变革的重要研究领域。目前,制取氢气的技术主要有三种:蒸汽甲烷重整、煤气化以及水的裂解[5-6],前两种方法在氢气的生产过程中会产生CO2,仍存在碳排放的问题。电催化技术分解水产生氢气是指在外加电流的作用下,利用催化剂的作用将水分解为纯氢和纯氧,是获取氢气的高效且绿色环保的技术之一,可实现无碳能源的循环利用。虽然在理论上实现水全分解所需要的过电位仅为1.23 V,但由于其具有多电子参与的复杂反应特征,动力学反应过程受到明显抑制,因而在实际应用中必须施加一个非常大的过电位以克服反应的动力学势垒,这就造成了大规模电解水过程中巨大的电能消耗,因此,开发高效的析氢催化剂对于降低能量消耗,实现电解水产氢技术的真正应用十分关键[7-8]。
铂系贵金属催化剂是目前对析氢反应具有最优催化能力的材料,但受制于其高昂的价格和资源的稀缺性,难以实现工业层面的大规模应用[9],因此,研制价格低廉、性能优异且稳定性好的非贵金属催化剂成为了目前研究的重点。在各种可能的催化材料中,二硫化钼因具有可调的电子结构、良好的催化稳定性以及丰富的储量,成为替代贵金属催化剂最具潜力的材料之一。然而,二硫化钼本身惰性基面的存在以及电导率较差的固有缺点严重限制了其析氢催化活性的提升。近年来,针对二硫化钼改性及其在电解水析氢领域研究人员已经开展了大量研究,并且取得了较为丰硕的成果,但其催化产氢性能与铂系贵金属催化剂相比仍有一定的差距。基于这一点,本文从电解水的机理及二硫化钼的结构特性出发,详细论述二硫化钼基催化剂的改性策略及其在催化析氢领域的最新研究进展,并针对未来的研究方向和发展趋势进行一定的展望,以期推动MoS2基析氢电催化剂早日实现工业化应用。
1 析氢反应(HER)机理
水的电解包含两个半反应,即发生在阴极的析氢反应(HER)和阳极的析氧反应(OER),此反应由van Troostwijik 和Deiman 于1789 年首次发现[10]。如图1(a)所示,HER 是一个两电子参与的多步反应过程,在酸性、碱性中的机理各不相同[11],酸性条件下的反应如式(1)~(3)所示[12]:
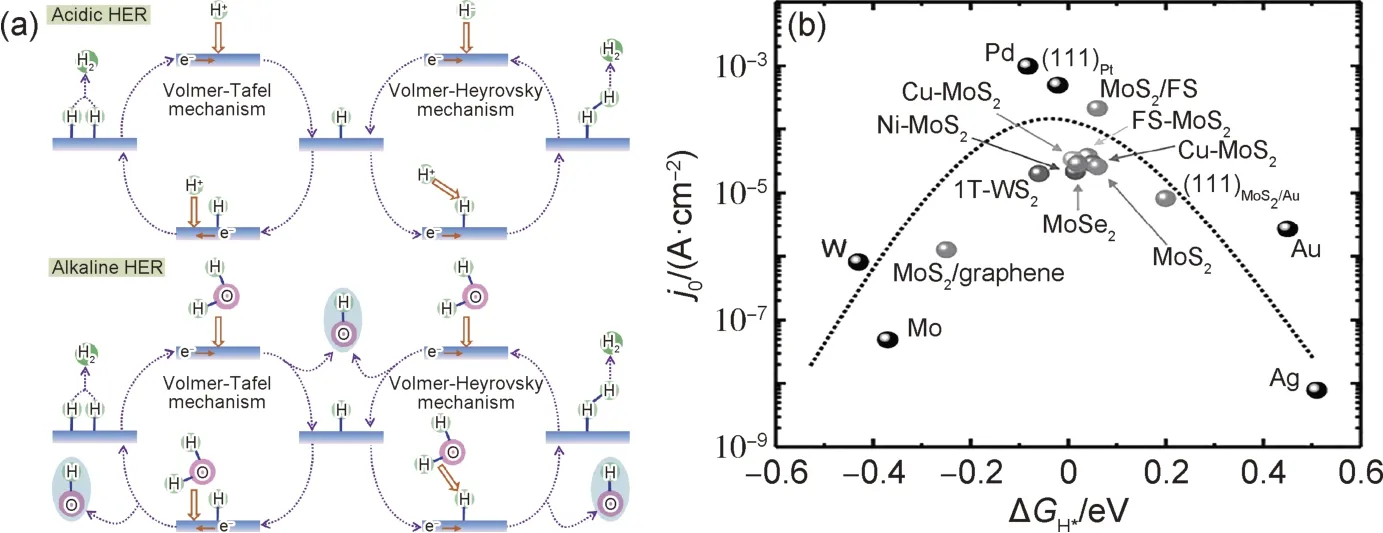
图1 催化析氢过程及不同催化剂催化活性(a)酸性及碱性条件下HER 路径[11];(b)电流密度与氢吸附能ΔGH*之间的关系[13]Fig.1 Catalytic hydrogen evolution process and catalytic activity of different catalysts(a)HER pathways in the acidic and alkaline media[11];(b)relationship between current density and hydrogen adsorption energy ΔGH*[13]
首先,H3O+得到一个电子并吸附在催化剂表面的活性位点,成为吸附氢(Hads),这一过程称为Volmer 过程(式(1))。吸附态的氢(Hads)随之与另一个H3O+耦合形成H2分子(Heyrovsky 过程,式(2)),或是直接与另一个表面吸附的氢结合形成H2(Tafel过程,式(3))。
碱性条件下的HER 过程如式(4)~(6)所示:
对于形成吸附氢的过程,主要是由H2O 取代H3O+进行还原反应生成吸附氢(Hads)。在接下来的Heyrovsky 过程中,也是由H2O 替代H3O+参与反应,并与吸附氢耦合形成H2分子;而在Tafel 过程中,同样是由两个Hads结合形成H2分子,这与在酸性条件下的反应一致。由于在碱性条件下的HER 过程在吸附氢的形成之前涉及水的解离过程,因此HER 在碱性条件下的反应过程更加复杂、动力学也更缓慢。
通过上述HER 反应机理可知,吸附氢在活性位点上的吸附能ΔGH*对于催化剂的性能起着至关重要的作用,它反映了H*与活性位点之间相互作用的强弱,如果吸附能ΔGH*为较大的负值,H*会牢固地吸附在催化剂表面,不利于后续的氢气脱附过程;相反,若ΔGH*为较大的正值,则H*很难吸附到活性位点处参与反应,从而影响整体HER 反应速率。由于不同材料对H*具有不同的ΔGH*数值,从而展现出不同的催化活性,其结果如图1(b)所示[13]。铂系贵金属催化剂具有绝对值最小的ΔGH*,更有利于H*的吸脱附过程,因而具有最为优异的HER 性能。MoS2基材料因对H*表现出适宜的吸附能,因此在HER 领域表现出了巨大应用潜力。
2 析氢电催化剂关键参数
用于评价析氢催化剂性能的参数主要包括过电位(η)、塔菲尔斜率(Tafel slope)以及稳定性等。通过对以上参数的分析,可以对催化剂性能优劣及寿命进行评价,从而判断其是否具有作为工业催化剂大规模使用的潜力。
2.1 过电位(η)
理论上水发生分解所需的电位为1.23 V,但在实际情况下由于活化能垒以及接触电阻和溶液内阻的存在,必须要施加一个比理论值高的电位来驱动水的分解过程,这部分高出的电位称为过电位(η)。通常采用电流密度为10 mA·cm-2时的过电位(η10)来作为评价催化剂催化活性的标准[12]。性能优异的催化剂能够在较低的过电位下达到10 mA·cm-2的电流密度。但应指出的是,由于受不同负载量及负载面积等因素的影响,且在工业化生产条件下往往需要大的实际电流密度,因此实验室条件下的η10不能作为评价催化剂性能优劣的唯一标准[14]。
2.2 塔菲尔斜率(Tafel slope)
塔菲尔斜率(Tafel slope)主要用于表征HER 的动力学特征,可通过Tafel 公式:η=a+blgj来拟合极化曲线数据得到具体数据,其中a为Tafel 截距,表示在零电流密度时的电极过电势值;j为电流密度;b为所求的塔菲尔斜率。较小的塔菲尔斜率意味着增加相同的电流密度时所需要的过电位更小,因此在HER过程中具有更快的动力学过程。此外,根据塔菲尔斜率的具体数据可对HER 过程中的速率决定步骤进行判断[15],从而对不同催化剂体系下的性能增强机理进行探索。
2.3 稳定性
相对于催化活性,长时间的稳定性是评价催化剂是否能实际应用的关键参数。稳定性的测试方法主要包括循环伏安(CV)测试或计时电位/电流测试[16]。在CV 测试中,经过数千次循环后的极化曲线的偏移量越小,说明催化剂的稳定性越好;而在计时电流法中,稳定性通常采用10 mA·cm-2电流密度下的电流密度-时间曲线来评估,稳定性较好的催化剂应在长时间的测试之后具有较小的电流密度损失[17]。
3 MoS2催化剂的结构特征
MoS2是二维过渡金属层状硫化物的典型代表,其结构如图2(a)[18]所示。层内是由交替排列的S 原子和Mo 原子组成,即两排S 原子中夹有一排Mo 原子,通过共价键形成三明治结构[19],而层间则是通过较弱的范德华力结合,从而形成类石墨垂直堆叠的六边形片状结构。MoS2存在多种晶型结构,主要有1T 型MoS2,2H 型MoS2和3R 型MoS2,其中以1T 相和2H相最为常见,它们的晶体结构如图2(b)所示[20]。在1T 相中,六个S 原子在一个Mo 原子周围形成八面体配位结构,而在2H 和3R 相中则是形成Mo 原子的三棱柱六配位结构。不同的配位结构使得MoS2的电子结构出现差异。对于1T 相MoS2,钼4d 轨道能级分裂为两组,两个自旋未配对的电子分别占据能量较低的两个简并轨道上,使得其表现出类金属性质,但1T 相MoS2不能稳定存在,一般由2H 相转变得来,且易在退火过程中发生相转变重新变回2H 相[21]。在2H 相MoS2中其4d 轨道能级则分裂为三组,其中自旋配对的两个电子占据能量较低的轨道,使得其呈现出半导体特性并且可以稳定存在[22],目前在电催化析氢领域的绝大部分研究工作都是围绕2H 相来进行。
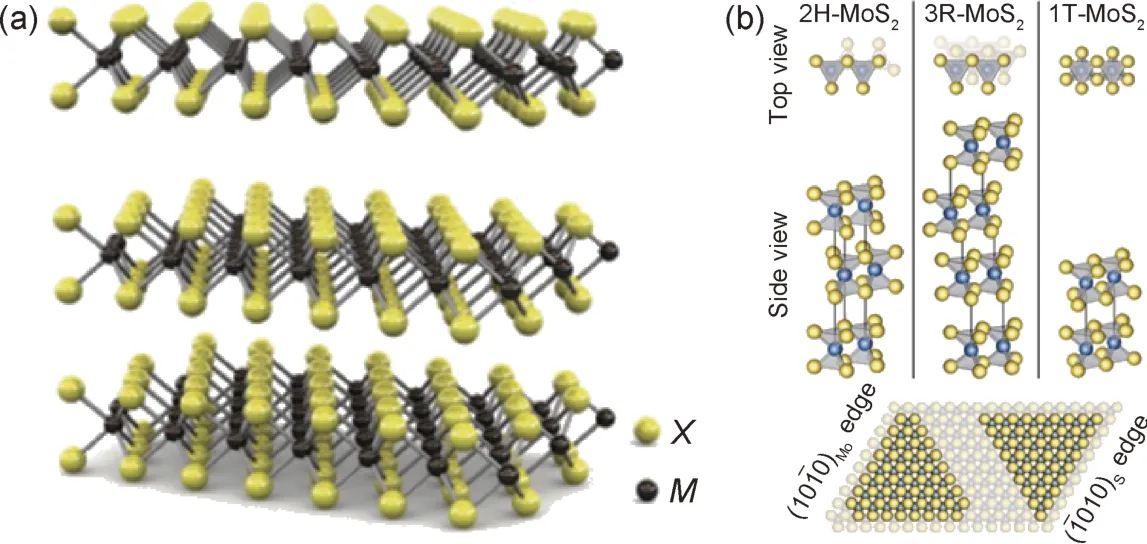
图2 层状过渡金属化合物及二硫化钼晶体结构(a)二维过渡金属硫化物结构[18];(b)2H,3R 和1T 晶体结构MoS2[20]Fig.2 Layered transition metal compounds and crystal structure of molybdenum disulfide(a)schematic diagram of the structure of two-dimensional transition metal sulfides[18];(b)structure of 2H, 3R and 1T polytypes of MoS2[20]
4 MoS2的改性策略
早期研究表明块体MoS2对电催化产氢几乎没有活性。直到2005 年,Hinnemann 等通过第一性原理(DFT)计算研究MoS2对H*的吸附势垒。结果表明(1010)面边缘的Mo 原子在H 原子与S 原子比例为1∶2 时的ΔGH为0.08 eV,与Pt 类似,并且接近理想值(0 eV)[23]。相反,MoS2基面在相同情况下的ΔGH数值却为1.92 eV,因此对HER 表现出惰性。这一结果表明,2H 相MoS2的催化性能主要来自其边缘位点,而基面的贡献则可以忽略[24]。此外,2H 相MoS2的半导体特性决定了其导电性较差。因此,活性位点的不足以及导电性较差是限制MoS2催化析氢性能提升的关键问题。针对这两个问题,研究人员采用了不同的策略来对其进行改性,主要是尽可能地暴露更多的边缘活性位点或是对惰性基面进行改性,从而创造新的活性位点;以及尽可能提高其导电性和载流子传输速度,从而提升催化产氢性能。目前发展的改性策略主要包括形貌调控、缺陷工程、界面工程及相转变等。
4.1 形貌调控
由于块体MoS2导电性差、活性位点少,导致其析氢反应动力学及电子转移过程较慢、催化活性较差。将MoS2电催化剂进行纳米化是最为可行的增加其比表面积和活性位点数量的方法。由于MoS2中活性位点主要位于边缘位点,因此对其进行形貌及尺寸的合理调控,从而得到富含边缘位点的MoS2纳米材料,最大化地暴露活性位点;同时比表面积的增加也能增强HER 过程中的电子/质子转移,加速气泡的脱附过程,从而有效增强HER 动力学过程,提升催化剂活性。截至目前,一维纳米线[25]、二维纳米片[26]、三维纳米颗粒[27]等不同形貌的纳米结构已经成功合成并应用于析氢催化领域。
Zhu 等[28]以NaCl 作为模板,通过低温化学气相沉积(low temperature chemical vapor deposition,LPCVD)技术制备了厚度可控的MoS2纳米片层材料,并且发现纳米片厚度对产氢性能具有很大的影响。厚度越小时,产物催化性能越好。当反应温度为550 ℃时得到的产物具有最佳性能,在10 mA·cm-2电流密度下的过电位为300 mV,且经1000 周次循环测试后没有发生明显衰减。Nguyen 等[29]通过简便的水热法制备出了具有不同形貌的MoS2纳米材料,并研究了其作为催化剂的产氢性能。结果表明,空心结构MoS2相对于纳米颗粒、纳米花、纳米球等形貌产物具有更为优异的HER 催化性能,在电流密度10 mA·cm-2时的过电位为230 mV,并且经过5×104s 的稳定性测试之后性能没有明显衰减。其原因在于空心结构具有最大的比表面积,从而使产物具有最大的电化学活性面积。Deng 等[30]通过模板加刻蚀方法合成了具有均匀介孔的三维泡沫结构MoS2,由于介孔周围定向垂直生长的MoS2纳米片增加了边缘活性位点的数量,且介孔结构的存在有效增加了电子和离子的传输速度,二者的协同作用有效促进产物催化性能的提升,仅需要210 mV 的过电位即可达到10 mA·cm-2的电流密度,并且具有良好的稳定性。这些研究表明合理设计MoS2的微观形貌对于提升其HER 性能非常有效。
除上述MoS2纳米材料被成功合成并应用于电催化析氢领域,近期研究人员合成了一系列MoS2量子点材料并探究了其电催化析氢性能。当MoS2的尺寸进一步减少后,其边缘活性位得以大量暴露并且有利于电子快速传输,从而有利于催化活性的提升[31]。Mohanty 等[32]通过简便的水热方法同时合成了MoS2量子点及MoS2纳米片。电化学测试结果表明MoS2量子点的催化性能相较于纳米片得到提升,其在酸性条件下10 mA·cm-2下的过电位为210 mV,并且在全pH范围内均具有优异的性能及稳定性。Huang 等[33]通过溶剂热方法将商用MoS2量子点负载于Ti3C2TxMXene 之上,MoS2量子点在MXene 基体之上均匀分布,理论计算结果表明复合催化剂具有良好的电导性以及适宜的ΔGH。得益于以上优点,复合催化剂的析氢起始电位仅为66 mV 且兼具长时间的催化稳定性。Peng 等[34]通过微波辅助方法将O 原子部分取代MoS2中的S 原子,从而制备了还原氧化石墨烯上的氧掺杂MoS2量子点,Raman 测试结果表明氧的成功掺杂。得益于量子点在rGO 上的均匀分布使得活性位点得以大量暴露以及氧掺杂效应,复合催化剂在酸性条件及10 mA·cm-2电流密度下的过电位仅为76 mV,并在1000 周次循环稳定性测试之后性能无明显衰减。以上研究结果表明MoS2量子点具有丰富的催化活性位点,催化性能较为优异,但如何保持其长时间的结构稳定性和催化稳定性仍是当前需要解决的难题。
4.2 缺陷工程
形貌调控是提高MoS2HER 动力学的有效途径,但其却受限于对HER 表现出惰性的MoS2基面,对于性能的提升作用有限。针对这一问题,研究者进一步发展了表面缺陷工程策略。通过缺陷的引入能够对MoS2的电子结构进行有效调节,改善惰性位点对氢的吸附自由能,进而增加了基面上的活性位点数量,显著增强MoS2基面的催化活性。此外还能改变材料的本征电导率,从热动力学的角度达到改善催化性能的目的。根据缺陷类型的不同可分为杂原子缺陷[35-37]、空位缺陷[38-41]及合金化[42-43]等,缺陷的引入方式包括可控生长[44-45]、后处理[46-47]、刻蚀[48-49]及机械剥离[50]等。
4.2.1 杂原子缺陷
元素掺杂是在MoS2中引入缺陷的常用手段,也是目前应用最为广泛的改性技术之一。通过在MoS2结构中形成掺杂能级,使掺杂轨道和分子轨道之间发生轨道杂化,进而调控产物电子结构和价带及导带的电位。因此,掺杂不但能够改变活性位点的数量,而且实现了本征电导率的有效调控,因而显著提升MoS2的电催化性能。目前研究的掺杂元素的种类主要包括金属和非金属,其中金属元素的掺杂研究主要集中在Co,Ni,Zn,V 等过渡金属[35,51-54]。非金属元素的掺杂主要为N,P,O 等元素[37,55-59]。两者的不同之处在于金属掺杂是通过金属原子替代Mo 原子来实现,而非金属掺杂是通过非金属原子取代S 原子或吸附于Mo 原子之上来实现[60]。
Ma 等[52]通过水热方法合成了石墨烯负载钴掺杂MoS2超薄纳米片(Co-MoS2/rGO),经过Co 掺杂的MoS2呈现出超薄的片层结构,且导电性得到一定提高。复合催化剂在10 mA·cm-2下的过电位为147 mV,并且在经过12 h 后的稳定性测试后没有出现明显的性能衰减(如图3(a)~(c)所示)。Wang 等[54]通过水热浸渍以及退火处理实现了单原子Ni 掺杂的MoS2。实验和理论结果表明Ni 原子倾向于分布在边缘S 位点以及由S,Mo 原子所形成的六边形平面的顶部位置,前者经过Ni 修饰后的ΔGH由-0.79 eV 变化为0.1 eV,而后者则由2.16 eV 变为0.8 eV。因此,通过Ni 修饰降低了基体对氢的吸附能垒,从而提高基体以及边缘位点位置活性位点的数量。近期研究表明,Zn 和V 掺杂对MoS2也具有很好的改性效果。其中Zn是通过提高能级位置以使电子能在更低的外加能量下进行传输,从而提高催化性能[53];而V 则可以提高MoS2的活性面积以及将费米能级向导带移动,从而增强HER 性能[35]。因此通过金属元素对MoS2掺杂形成外来杂原子缺陷,可以有效改变其物理化学性质,调节催化剂对于氢的吸附过程,从而增强催化活性。
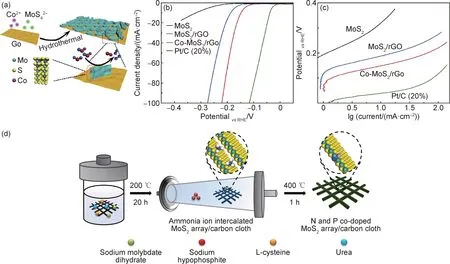
图3 MoS2元素掺杂工程(a)Co-MoS2/rGO 合成过程[52];(b),(c)HER 性能[52];(d)氮、磷共掺MoS2合成过程[57]Fig.3 MoS2 element doping engineering(a)fabrication process of Co-MoS2/rGO[52];(b),(c)HER performance[52];(d)synthesis process of nitrogen and phosphorus co-doped MoS2[57]
通过金属元素对MoS2进行掺杂处理可以增强其导电性、引入额外的活性位点,从而有效增强HER 性能,但金属掺杂也存在着明显的缺点。由于物性差异,大原子尺寸的金属离子很难大量掺杂进入MoS2的基体中,并且金属的掺杂还易引起MoS2结构的不稳定,反而容易造成性能的下降[61]。因此,研究者进一步尝试利用非金属(N,P,O 等)掺杂来提高MoS2的催化析氢性能。例如,Xiao 等[58]通过一步烧结法制备了N 掺杂的MoS2催化剂,其在100 mA·cm-2大电流密度下的过电位仅为121 mV,并且具有极小的Tafel斜率(41 mV·dec-1)。第一性原理计算表明N 原子倾向于取代MoS2边缘位点和(001)面的S 原子,从而有效调节了掺杂位点附近的S 原子对于H 的吸附能;并且由于费米能级附近Mo3d-S2p-N2p 的强杂化作用,引起电荷在N 掺杂基面上的扩散,从而增强了MoS2基体的电导率,有效促进了电荷的快速传输,两者的协同作用共同提高了MoS2的催化性能。Wu 等[37]通过静电纺丝技术合成了碳纳米纤维负载的P 掺杂MoS2二维纳米片,在具有优异HER 性能的同时兼具长时间的稳定性。实验及理论计算结果表明,P 掺杂不但减小了MoS2半导体的带隙,增加其电导率;并且通过改变MoS2的晶面间距降低对H 的吸附能垒[37,56]。在单元素掺杂的基础上,研究者进一步探索了多元素掺杂对性能的促进作用。例如Sun 等[57]通过水热和热处理在碳布(CC)上负载了N,P 共掺的MoS2(如图3(d)所示)。其碱性条件下的η10仅为78 mV,且Tafel 斜率为58.4 mV·dec-1,表现出优异的催化产氢性能。研究结果表明,由于多元素的协同作用,位于MoS2基体表面的Mo-N-P 位点不但具有优异的氢吸附性能,而且兼具较高的键合强度,因此增强了其HER 的动力学过程以及电化学稳定性。不同元素掺杂对MoS2材料析氢催化性能作用的详细对比如表1所示[35,37,52-59,62-63]。
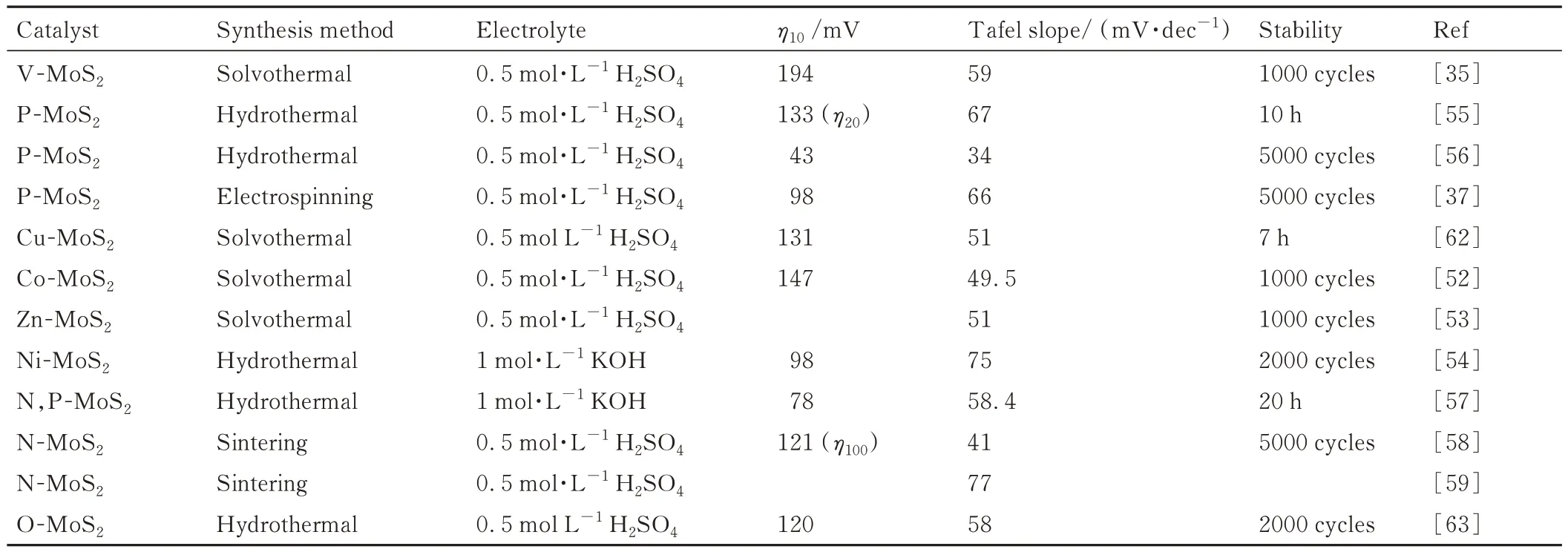
表1 元素掺杂MoS2 催化剂的HER 性能对比Table 1 HER performance of element-doped MoS2 electrocatalysts
4.2.2 合金化
通过元素掺杂可以有效调节MoS2结构内的局域电子结构,进而增加MoS2中活性位点的数量以提升HER 性能。然而受限于掺杂元素与主体元素物理和化学性质的不同,难以将掺杂原子以高浓度掺杂进MoS2中,限制了电子结构的连续化调节。为了解决这一问题,研究者进一步发展了合金化的改性策略。合金化主要是利用与Mo,S 同族的W 以及Se 进行掺杂,因而可以大量取代MoS2中的Mo 原子或S 原子,且合金化还可以改变材料的物理和化学性质、增强其电催化性能[43]。Wang 等[42]通过高温热处理偏钨酸铵、钼酸铵以及硫脲的混合物制备了Mo1-xWxS2合金,x值可以通过控制偏钨酸铵和钼酸钠之间的比例来调控。理论计算表明:当x=0.5 时材料对活性氢的吸附自由能最低,且具有最佳的电荷迁移能力,因而表现出最优异的HER 催化性能。酸性条件下的η10仅为138 mV,且经过2000 周次CV 循环后仍具有较好的稳定性。Gong 等[64]制备了超薄MoS2(1-x)Se2x合金纳米薄片,结果表明Se 的引入可以对Mo 的d 带电子结构进行连续调节,通过减小ΔGH实现HER 性能的有效增强。
4.2.3 空位缺陷
除了边缘活性位点,MoS2基面中的S 空位也被证明是析氢反应的活性中心。半导体MoS2中存在S 空位时,其相当于n 型掺杂,起到电子供体的作用,使得费米能级附近的电子态密度增加,从而改变了MoS2的电子和能带结构,进而改善其导电性和对于氢的吸附过程,达到提升催化性能的目的。常用于引入S 空位的方法有等离子刻蚀[65]、湿法刻蚀[66]、激光刻蚀[67]、辐照处理[68]、氢气退火[40]以及电化学脱硫[69]等,通过以上方法可以将MoS2中的部分S 原子去除,从而形成S 空位。
例如,Dong 等[45]通过CVD 方法合成了具有丰富S 空位的单层MoS2六方薄片。电化学测试结果表明在引入S 空位后,其性能较原始MoS2得到大幅提升。DFT 计算表明当S 空位引入单层MoS2中时形成局部能级,由S 空位贡献的多余电荷载流子在室温热激活作用下占据较低的导带能级,使得材料电导率提高。因此,通过引入S 空位不仅可以增加活性位点的数量,还可以提高MoS2的本征电导率,从而协同提高催化活性。Chen 等[44]通过微波辅助溶剂热法制备了MoS2/rGO 复合材料。由于其合成过程具有快速和低温等特点,因此制备的MoS2中含有丰富的S空位缺陷,从而为析氢反应提供了更多的催化活性位点。此外rGO 和MoS2颗粒之间的相互作用促进了电荷的转移过程,共同促进电催化性能的提升,这一工作为合成缺陷丰富的MoS2基催化剂提供了新的策略。
S 空位的数量及其在MoS2结构中的不同状态也会对其催化性能产生显著影响。Li 等[39]通过模拟计算研究了S 空位含量对于HER 性能的影响,S 空位的引入使得氢在暴露的Mo 原子上的吸附能明显降低,这表明MoS2基面的HER 性能得到有效改善。且当S空位的含量在9%~19%(相对应于其数量的S 原子被去除)时,其氢吸附自由能与边缘位点相当,具有最优的催化活性。通过电子及能带结构的计算对其性能提升的原因进行了进一步解释。首先,S 空位的引入会引起费米能级附近新缺陷能级的出现,即S 空位附近Kohn-Sham 轨道的形成。且随着S 空位数量的增加,能带会逐渐向费米能级移动,造成间隙态密度的增加,从而增强对氢的吸附作用,因而S 空位数量存在最优值。Zhou 等[41]从H 原子S 轨道和相邻原子的价轨道的局域态密度角度解释了MoS2中引入空位后对氢吸附的影响。在存在S 空位的情况下,H 原子S 轨道和相邻原子(Mo 和S)的价轨道的局域态密度在相同的能量位置并没有同时出现杂化峰,因此H2分子与其相邻的S 原子和Mo 原子之间的相互作用均较弱,证实H2分子可以轻易从材料表面脱附。此外当1T 相MoS2中存在S 空位时,作为HER 控速步骤的Volmer反应能垒为1.21 eV,高于原始1T 相,这表明在1T 相MoS2中S 空位的存在反而会对HER 性能产生不利的影响。因此,针对不同物相结构的硫化钼,需要结合理论方面的深入认知并加以对缺陷结构的有效调控来实现HER 性能的有效增强,这一点仍需要加以深入研究。
5 界面工程
界面工程是指MoS2负载于基体材料之上或与其他材料形成异质结构,借助于MoS2与基体材料或其他材料之间所形成的界面处独特的物理化学特性,从而达到调节其整体催化性能的目的[70-71]。
将MoS2纳米材料负载于导电性良好的基体之上,不仅可以防止其在催化过程中由于团聚所导致的性能衰减,更可以增加材料整体导电率,促进电子传输过程,从而加快反应速度。Wu 等[72]通过简单快速的溶剂热方法将MoS2纳米片负载于乙炔黑之上,乙炔黑的存在不仅可以使催化剂暴露出更多的活性位点,更可以提高单位时间内单个活性位点的转换数,有效提高催化速率。Cao 等[73]通过水热加硫化处理制备了MoS2@CoS2/Graphene 析氢催化剂,研究表明CoS2的引入和硫化处理有利于1T 相MoS2的生成进而提升其导电性,石墨烯基体良好的导电率更有利于电荷之间的快速转移。该催化剂在呈现出优异的催化活性的同时,在酸性电解液中表现出优异的稳定性。近期,研究者们构建了新颖的基体/MoS2复合材料或异质结构用于提升其析氢催化性能。Zeleke 等[74]将单分子MoS2负载于炭化后的聚丙烯氰上,扩展X 射线吸收精细结构结果排除了钼-钼之间相互作用,从而确定MoS2处于相互分离的单分子状态,TEM 分析进一步确定其分子尺寸约为1.31 nm。得益于Mo 原子的低配位、4d 轨道的高空位以及单分子表面的最大化利用,其HER 性能相较于块体MoS2材料得以大幅提升。Tong 等[75]创造性地使用天然管状柳絮作为模板,合成了碳微管上原位负载MoS2的杂化物结构,一方面碳微管上原位生长的MoS2薄片有利于暴露更多的活性位点,另一方面碳微管可以作为电子快速传输的通道。综合以上优点,此杂化物呈现出优异的析氢催化性能并表现出可观的稳定性。
碱性条件下电解水是实现大规模制氢的重要途径之一,然而其实现需要具有大电流密度以及超长稳定性的催化剂做支撑。将MoS2与其他电催化材料构建成具有异质结构的复合电催化剂是解决上述问题的有效措施。在异质界面处电子会发生重排从而导致其对于氢的吸附能发生变化,进而调节析氢催化性能。此外由于不同催化剂对于析氢、析氧两个半反应的活性不同,因此异质结构的构建有望将MoS2用作双功能催化剂,从而实现真正意义上的大规模工业化制氢[76]。近期,Wu 等[77]设计了一种基于氮化镍和单斜二硫化钼异质结构的高效电解水催化剂。2MMoS2中的活性Mo—Mo 键可以作为电子的供体进而调节Ni 原子和N 原子在费米能级附近的活性电子状态,进而加强催化性能。由于碱性条件下HER 过程涉及水的分解以及氢的吸附两个连续的过程,而Ni3N 中的Ni 位点对水分解呈现出较小的能量势垒,进而为HER 过程提供更多的氢源作为反应物,加速产氢过程的进行。此外,异质结构的形成使得催化剂具有更好的电子传导特性。基于以上优点,此催化剂在1000 mA·cm-2的高电流密度下全解水电池电压仅为1.644 V,且持续300 h 后性能几乎没有衰减。Zhang等[78]通过原位自硫化策略合成了具有双轴应变效应的Ni3S2@MoS2核壳结构催化剂,其外部的MoS2壳层可以在1~5 层的范围内精确调控,当壳层为两层时,碱性条件下其性能达到最佳,10 mA·cm-2下的过电位仅为78.1 mV,并且在经过80 h 后的稳定性测试后性能没有明显衰减。其优异的HER 性能受到应变、NiS2/MoS2之间电子转移、S 空位及MoS2壳层数目的共同作用。
6 相转变
1T 相MoS2中,Mo 的4d 轨道分裂为三重简并轨道,其价电子只占据其中两个轨道,因而有利于基面上的S 原子与H 之间形成杂化轨道,从而使得1T 相与2H 半导体相相比具有更多的活性位点,并且使其展现出金属特性,具有更好的导电性,因此1T 相MoS2具有更高的HER 活性[79-80]。虽然碱金属嵌入-剥离、Ar等离子体处理、机械应变以及电子束辐照等方法已经成功地用于MoS2相转变过程,并且成功制备非稳态1T 相MoS2。但上述方法的效率相当低,限制了其进一步的应用[81]。近来,通过简单的溶剂热方法进行1T相MoS2的制备得到了广泛的研究,并取得了很大的进展[62,82-88]。例如,Wang 等[88]以钼酸铵、硫代乙酰胺、碳酸氢铵为原料,通过简单的溶剂热方法合成了高活性且稳定的复相催化剂(1T/2H MoS2),通过对不同类型S 原子数目百分比进行XPS 半定量分析表明1T相MoS2质量分数为61.5%。产物在酸性条件下10 mA·cm-2电流密度下的过电位为234 mV,并经1000 周次CV 循环后测试仍保持良好催化活性。Geng 等[84]以三氧化钼、尿素、硫代乙酰胺为原料通过简单的溶剂热方法合成了纯1T 相MoS2,并发现产物的电荷转移阻抗仅为半导体相的1/50。酸性条件下,催化剂在10 mA·cm-2下的过电位仅为175 mV,Tafel斜率为42 mV/dec,并具有优异的稳定性。
近期研究表明,通过元素掺杂不但可以促进2H相MoS2向1T 相转变,而且可以进一步调节材料的电子结构,协同增强HER 性能。Jiang 等[85]通过溶剂热方法合成了Se,O 共掺的MoS2。结果表明Se 和O 的掺杂不仅诱导了2H 相向1T 相进行转变,而且在MoS2基面中产生S 空位,导电性的增强以及活性位点数量的提升使得HER 性能得以大幅度提高。Gao 等[83]通过磁控溅射的方法在石墨烯上合成了C 掺杂的1T-2H 混合相MoS2纳米片,其中1T 相占比达到60%。并通过Raman 和XPS 进一步研究了其相变过程,发现离子轰击和C 掺杂造成的S 原子层滑移是导致2H 相MoS2向1T 相转变的主要原因。产物在碱性条件下的η10仅为40 mV,并具有出色的稳定性。Li 等[89]通过电位循环法将Pt 掺入MoS2的晶格之中并实现了部分MoS22H 相向1T 相的转变,结果如图4(a)所示。通过Pt@MoS2的STEM 照片可以清晰地分辨出2H 相及1T 相MoS2的存在,形成能计算表明Pt 更易掺杂进入1T 相MoS2的晶格之中的S 空位位置,且相对于其他位置其更有利于促进HER 性能的提升(如图4(b)所示)。Sun 等[90]进一步研究了N, Pt 共掺MoS2材料的制备及催化性能,发现N2等离子体处理有利于Pt 在MoS2中的掺杂,并有效促进了1T 相MoS2的转化率(87%)。由于1T 相的存在及元素掺杂的协同效应,使得产物的HER 性能得到了巨幅提升。10 mA·cm-2电流密度下的过电位仅为38 mV,且实现了1000 h 的长时间稳定运行。精细结构表征及理论计算结果证实掺杂能够有效调节MoS2的电子及配位结构,从而激活基体表面的S 位点,同时改变了水解过程中过渡态的能量势垒,形成了有利于水吸附和解吸的空spz轨道(图4),进而有效增强了MoS2的催化性能。这些结果表明在对MoS2进行改性时,采用多种策略有效结合,通过相互间的协同作用更有利于MoS2催化性能的提升。
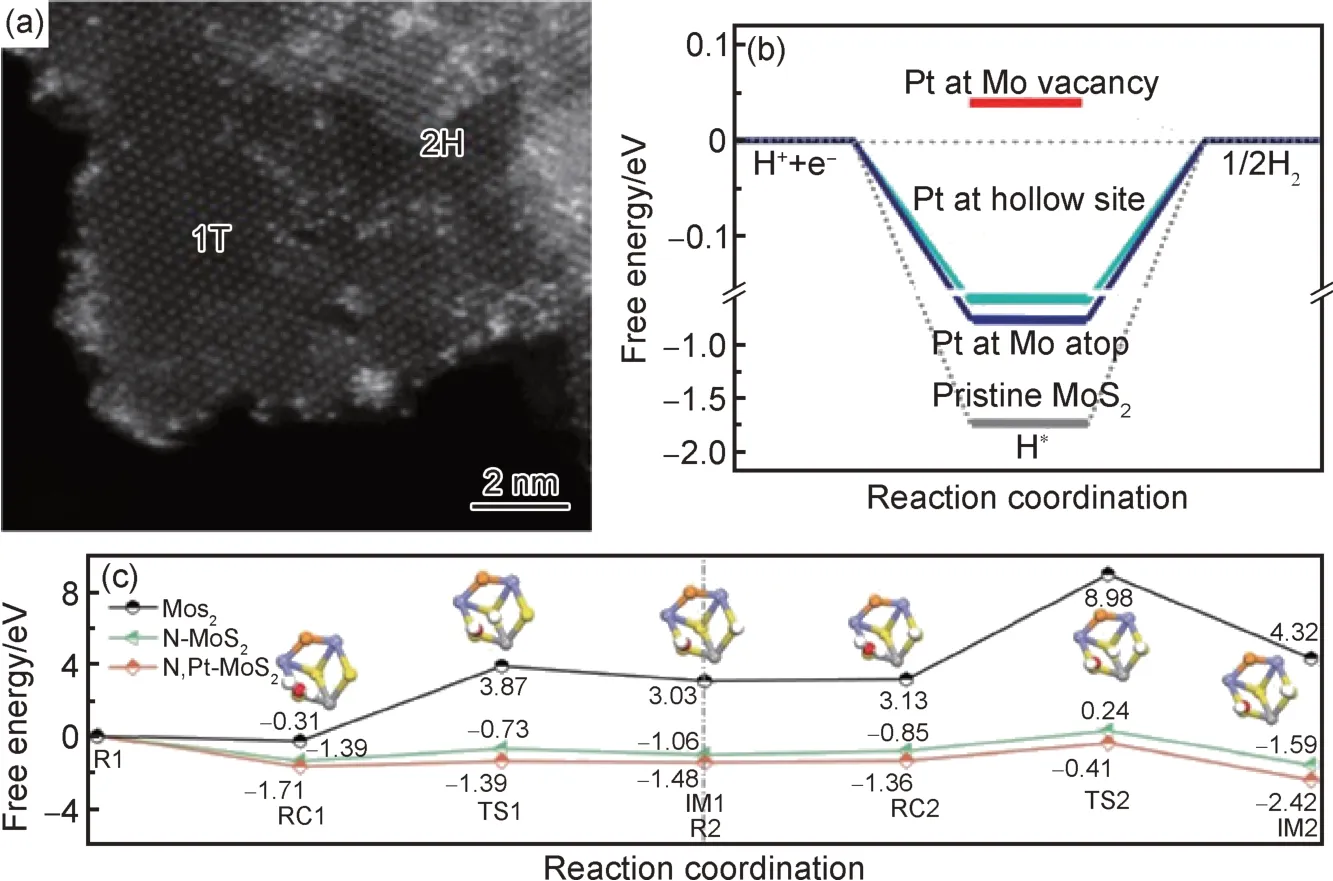
图4 元素掺杂MoS2及催化性能变化(a)Pt@MoS2的STEM 照片[89];(b)不同构型的Pt@MoS2在平衡电位下HER 过程自由能变化[89];(c)不同掺杂类型的MoS2 HER 过程的自由能变化[90]Fig.4 Element doped MoS2 and the change of catalytic performance(a)STEM of Pt@MoS2[89];(b)free energy profiles of HER at the equilibrium potential over different configurations of Pt@MoS2[89];(c)free energy changes in MoS2 HER processes with different doping types[90]
7 结束语
本文由电解水的机理出发,综述了MoS2基催化剂改性及其在电催化产氢领域的最新研究进展。针对制约MoS2基析氢电催化剂性能提升的惰性基面以及导电性较差的内在原因,论述了形貌调控、缺陷工程、界面工程及相转变等增强HER 性能的改性措施,并对其性能提升机理进行了重点分析。形貌工程可以将MoS2边缘活性位点最大化地进行暴露;缺陷工程、界面工程可以调节MoS2的电子和能带结构,改变硫、钼原子的配位结构,从而改善其对于氢的吸脱附过程,加快电子传输过程,增强催化性能。通过相转变获得1T 相MoS2则可以大幅提升其本征催化活性。通过分析进一步指出将不同改性策略进行结合,更有利于通过其协同作用有效增强MoS2的催化性能。
尽管目前MoS2基电催化剂的研究已经取得了诸多进展,但仍存在一些不足之处。首先,催化性能与贵金属Pt 相比仍有一定差距,实现工业化应用仍有一段很长的路要走;其次,尽管目前发展了许多不同的方法用于MoS2结构的调控,但是在原子尺度上对MoS2基催化剂的缺陷与成分进行精确调控的手段仍有待于进一步发展;此外,对于催化机理的理解仍不够深入,无法从理论的角度对催化剂的设计及合成进行指导,限制了性能的进一步提升。要解决这些问题,需要在后续的研究中重点关注以下方面:首先是加强电催化过程的理论研究,借助先进的理论计算方法,模拟分析电催化产氢的基本原理,并结合原位分析技术,对电催化产氢过程进行有效解析,在建立内在显微构型与表观催化性能关联机制的基础上,为材料的设计与合成提供理论指导;其次,需要进一步发展多要素协同的改性策略,从原子尺度实现材料表观成分及显微结构的有效调控,尽可能地实现活性位点数量及导电性的协同提升;此外,还需要对制备技术加以深入研究,力求通过简单易行的方式实现MoS2催化剂的有效合成,为低成本、高性能的工业化规模应用提供材料基础。
猜你喜欢
食品安全导刊(2021年20期)2021-11-28
陶瓷学报(2019年5期)2019-01-12
电镀与环保(2017年5期)2017-12-19
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年2期)2017-01-20
电镀与环保(2016年2期)2017-01-20
现代工业经济和信息化(2016年12期)2016-05-17
读者欣赏(2014年6期)2014-07-03
语文知识(2014年2期)2014-02-28
