
用生物信息学方法分析鉴定肺腺癌中的hub 基因
2023-12-16 07:28:20吕金海
怀化学院学报 2023年5期
吕金海
(怀化学院,湖南 怀化 418008)
肺癌是常见的癌症类型之一[1],其分为小细胞肺癌(Small Cell LungCancer,SCLC)和非小细胞肺癌(Nonsmall Cell LungCancer,NSCLC)2 种类型。其中,NSCLC约占肺癌的85%[2]。肺腺癌(LungAdenocarcinoma,LUAD)是非小细胞肺癌的主要亚型之一。肺癌患者早期无明显症状,大部分患者被发现时已经处于中晚期[3]。肺腺癌与多种风险因素有关,其成因与发病机制尚不完全清楚。
在癌症基因组图谱(The Cancer Genome Atlas,TCGA)、国际癌症基因组联盟(International Cancer Genome Consortium,ICGC)等大样本肿瘤研究项目开展后,肿瘤样本中生成了大量对肿瘤研究有帮助的数据[4-5]。此外,一些小规模癌症项目的数据也被存入高通量基因表达数据库(Gene Expression Omnibus,GEO)等数据库[6]中,为生物信息学分析提供了一些帮助。
在各种实验技术都存在局限性的情况下[7],基因芯片技术和高通量测序技术的应用给予了癌症研究一个新视角。公共数据库中的大量数据为不同类型癌症基因表达的综合分析提供了便利。最近,一些研究人员将生物信息学得到的结果与临床数据相结合,发现了用于不同类型癌症的诊断、治疗和预后的新的生物标志物[8-10],这类新的生物标志物被称为hub 基因。基于此,CHEN 等人[11]发现了4 个有望作为生物标记物的hub 基因。GUO 等人[12]鉴定出了9 个与肾上腺皮质癌相关的hub 基因并报道了相关通路。
本研究主要是利用综合生物信息学探究癌症的底层发病原理,并寻找其中可能的分子作用机制来改进诊断方法和预防措施。我们从GEO 中获取了GSE116959[13]和GSE118370[14]2 张基因芯片,限定log2FC值大于1.00 且p值小于5.00×10-2,得到了差异表达基因(Differentially Expressed Genes,DEGs),并将其进行基因本体论(Gene Ontology,GO)和基因组的京都百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)富集分析。此后,我们组建了完整的差异表达基因的蛋白质-蛋白质互作(Protein-protein Interaction,PPI)网络和加权基因共表达网络(Weighted Gene Coexpression Network Analysis,WGCNA)。本文利用一系列生物信息学方法,确定了hub 基因,并使用富集分析来确定与肺腺癌相关的关键通路。我们进行了生存分析来探究hub 基因的表达与肺腺癌预后的关系。本研究使用了多个平台的数据进行综合分析,大大提高了hub 基因的可靠性。本研究结果可从分子水平进一步了解肺腺癌的发生发展,为肺腺癌的诊断、治疗、监测和预后提供一些准确、有实用价值的标志物。
1 材料与方法
1.1 数据来源
GEO 数据库是由美国国家生物技术信息中心(National Center for Biotechnology Information,NCBI)于2000 年创立的公共基因表达数据库。我们通过GEO数据库评估后选择并下载了2 个基因表达谱数据集:GSE116959 和GSE118370,其中GSE116959 含有68个样本(57 个肺腺癌样本和11 个正常对照样本),GSE118370 含有12 个样本(6 个肺腺癌样本和6 个正常对照样本)。
1.2 方法
1.2.1 批次效应的消除和DEGs 筛选
在高通量实验中,异构性和潜在变量被认为是偏差和变异性的主要来源。基因组实验中最著名的潜在变异来源是批次效应。批次效应的影响可能很严重,甚至完全损害生物学结果。
我们首先将GEO 中得到的数据使用R 包“tidyverse”中的“left_join”命令进行合并,然后使用R包“sva”中的“ComBat”函数消除批次效应后,再选用R 包“limma”[15]比对肺腺癌组织与正常组织的数据,最后筛选出矫正后P<5.00×10-2的数据,其中上调基因设置为“log2FC>1.00”,下调基因设置为“log2FC<-1.00”。
1.2.2 DEGs 的GO 分析和KEGG 分析
转录组学和蛋白质组学生成的数据越来越多,我们需要采用综合策略进行分析。GO 数据库将基因与该数据库系统中的生物学术语紧密联系,并把基因功能分为3 个部分:细胞组分(Cellular Component,CC)、分子功能(Molecular Function,MF)、生物过程(Biological Process,BP)[16]。KEGG 是一个包含基因表达途径和功能的数据库,旨在通过基因组信息和生物信息学来预测细胞中复杂的通路及生物学行为[17]。R 包“clusterProfiler”支持3 种物种,包括人类、小鼠和酵母。它提供了一种基因分类方法:groupGO,并可以自动进行基因簇富集分析[18]。我们对获得的DEGs进行GO分析和KEGG 富集分析,并设置P=5.00×10-2为筛选阈值。
1.2.3 PPI 网络构建与分析
我们首先将筛选出的DEGs 输入在线数据库检索相互作用基因/蛋白质的搜索工具(Search Tool for the Retrieval of Interacting Genes/Proteins,STRING)数据库[19](https://www.string-db.org),然后对Cytoscape[20](https://cytoscape.org)注释和分析得到的PPI 网络使用cytoHubba[21]插件进行hub 基因的筛选,最后通过MCODE[22]插件识别PPI 网络中最重要的模块。
1.2.4 加权基因共表达网络的构建
WGCNA 是一种系统生物学方法,用于描述微阵列样品中基因之间的相关模式。WGCNA 软件包包含一组全面的功能,用于对大型高维数据集进行相关网络分析,其使用无监督聚类来识别基因模块,我们使用R 包“WGCNA”[23]构建加权基因共表达网络。
软阈值是基于近似无尺度网络的一种准则,同时也是可以减少误差的一种方式。它使得构建的网络更符合幂律分布和无尺度网络特征,并使得结果更具有生物数据特征。本文首先通过pick Soft Threshold 函数计算出软阈值12,紧接着通过该软阈值构建加权基因共表达网络,然后对基因进行聚类后使用动态剪树法识别基因模块合并,其次将相似的基因聚类到相同颜色的模块中,最后计算皮尔森相关系数以评估模块与表型之间的潜在关联性。
1.2.5 基于Oncomine 数据库的Meta 分析
微阵列研究的一个问题是假阳性结果很普遍,而Meta 分析可以从各种各样的微阵列数据集中识别和评估多个基因表达特征的交集,从而真实反映肿瘤的基本转录特征。
赛默飞公司开发了商业化的Oncomine 数据库[24],该数据库是目前世界上最大的癌基因芯片数据库和整合数据挖掘平台。用户可以使用Oncomine 选择适当的研究进行比较,然后通过Meta 分析来确定多个独立研究中显著过表达或低表达的基因。本研究通过Oncomine 在线数据库分析筛选出的hub 基因在肺腺癌组织与正常组织中的表达情况来验证hub 基因的可靠性。
1.2.6 生存分析
2017 年上线的GEPIAWeb 是其基于TCGA 和基因型-组织表达(Genotype-Tissue expression,GTex)的服务器。GEPIA2(http://GEPIA.Cancer-pku.cn)是GEPIAWeb经过更新和增强的版本,可提供更多功能[25]。GEPIA2 可根据基因表达水平进行生存分析,该功能允许用户筛选基因在不同癌症类型中的预后影响。为了进一步评估hub 基因在本研究中的预后重要性,我们在此平台上进行了总体生存分析,用来检验每个基因的表达与总生存率之间的关系。
2 结果
2.1 DEGs 筛选
本文将GSE116959 与GSE118370 的数据进行集成并消除批次效应后,设置|log2FC|>1(log2FC=log2病人平均表达量/正常人平均表达量),校正后p<0.05(p大于0.05,被认为没有统计学意义),筛选得到1433 个差异基因后对差异基因的表达模式进行了研究,图1 显示上调的523 个差异基因,下调的910 个差异基因。
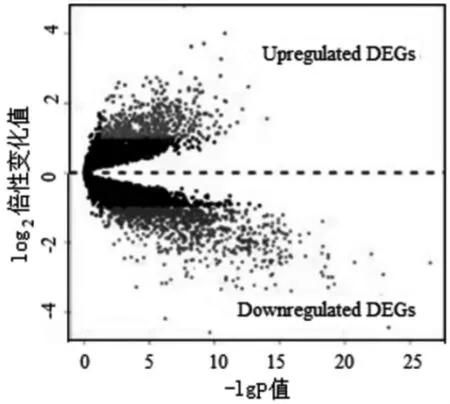
图1 差异基因火山图
2.2 GO 分析与KEGG 分析
对筛选出来的差异基因做GO 分析和KEGG 分析。GO 分析的结果显示,BP 主要围绕细胞外基质组织、细胞外结构组织和有丝分裂的细胞核分裂;CC 主要表现在含胶原的细胞外基质、分泌颗粒内腔和胶原三聚体等结构;MF 主要体现在细胞外基质结构组成、糖胺聚糖结合和肽聚合。KEGG 分析显示,差异基因主要富集在补体和凝血级联、细胞外基质(Extracellular matrix,ECM)受体相互作用和黏着作用的通路中,具体见表1。

表1 富集分析结果表
2.3 蛋白质互作网络构建与分析
使用STRING 分析DEGs 间的相互作用。结果显示共有716 个DEGs 网络节点和3759 条边被用于构建PPI 网络。图2 为使用cytoHubba 中的Degree算法初步筛选出20 个hub 基因:CDK1、CDC20、CCNA2、CCNB1、KIF11、CCNB2、KIF20A、KIF2C、GNG11、BUB1、FPR2、TOP2A、CENPE、PIK3R1、CDCA8、BUB1B、UBE2C、MAD2L1、AGT、ASPM。MCODE 可检测可能代表分子复合物的大型PPI 网络中的密集连接区域。该方法通过局部邻域密度和从局部密集的种子蛋白向外遍历的顶点加权,根据给定的参数隔离密集的区域。图3为使用MCODE 鉴定出了3 个最重要的基因簇。

图2 hub 基因网络图
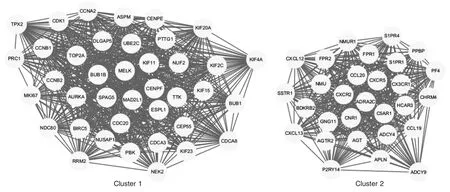
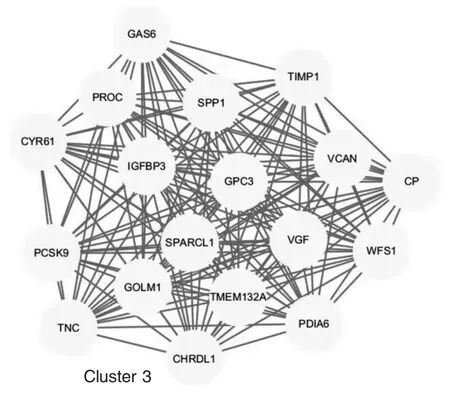
图3 MCODE 模块图
2.4 加权基因共表达网络的构建与分析
本文以12 为软阈值构建网络模块,通过对模块的进一步分析,计算模块间的差异,并构建基因模块树状图(见图4)。本文使用动态剪切树法识别基因模块,并将相似度较高的模块合并,最终得到18 个模块(见图5)。18 个不同带状矩形框代表18 个基因集模块。图4 显示树状图上的每一个峰都对应1 个基因,相似的基因被聚类到相同带状矩形框的模块中。表2显示数据集分为肺腺癌和正常2 种表型。相关系数大于0.6 的模块与性状显著相关。

表2 模块-性状相关性表

图4 基因模块树状图
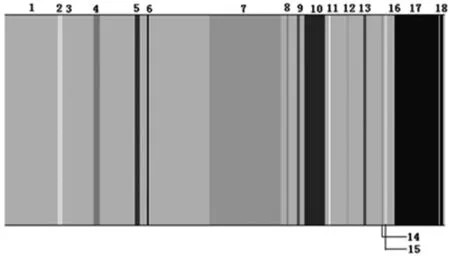
图5 基因矩形模块图
综合WGCNA 的基因模块树状图、基因矩形模块图、模块-性状相关性表和PPI 网络分析的结果,我们初步筛选出17 个hub 基因:CDK1、CDC20、CCNA2、CCNB1、KIF11、CCNB2、KIF20A、KIF2C、BUB1、TOP2A、CENPE、PIK3R1、CDCA8、BUB1B、UBE2C、MAD2L1、ASPM,并进行了进一步的分析和验证。
2.5 Oncomine 数据库差异表达分析
使用Oncomine 数据库,设置Analysis Type:Cancer vs.NormalAnalysis;Cancer Type:LungCancer;DataType:mRNA 选取肺腺癌患者和正常人的数据进行meta 分析,详细数据见表3。除没有研究发现PIK3R1 在肺腺癌患者与正常人中的表达量有差异外,所有hub 基因在肺腺癌与正常人中的表达量都具有统计学差异(p<0.05)。
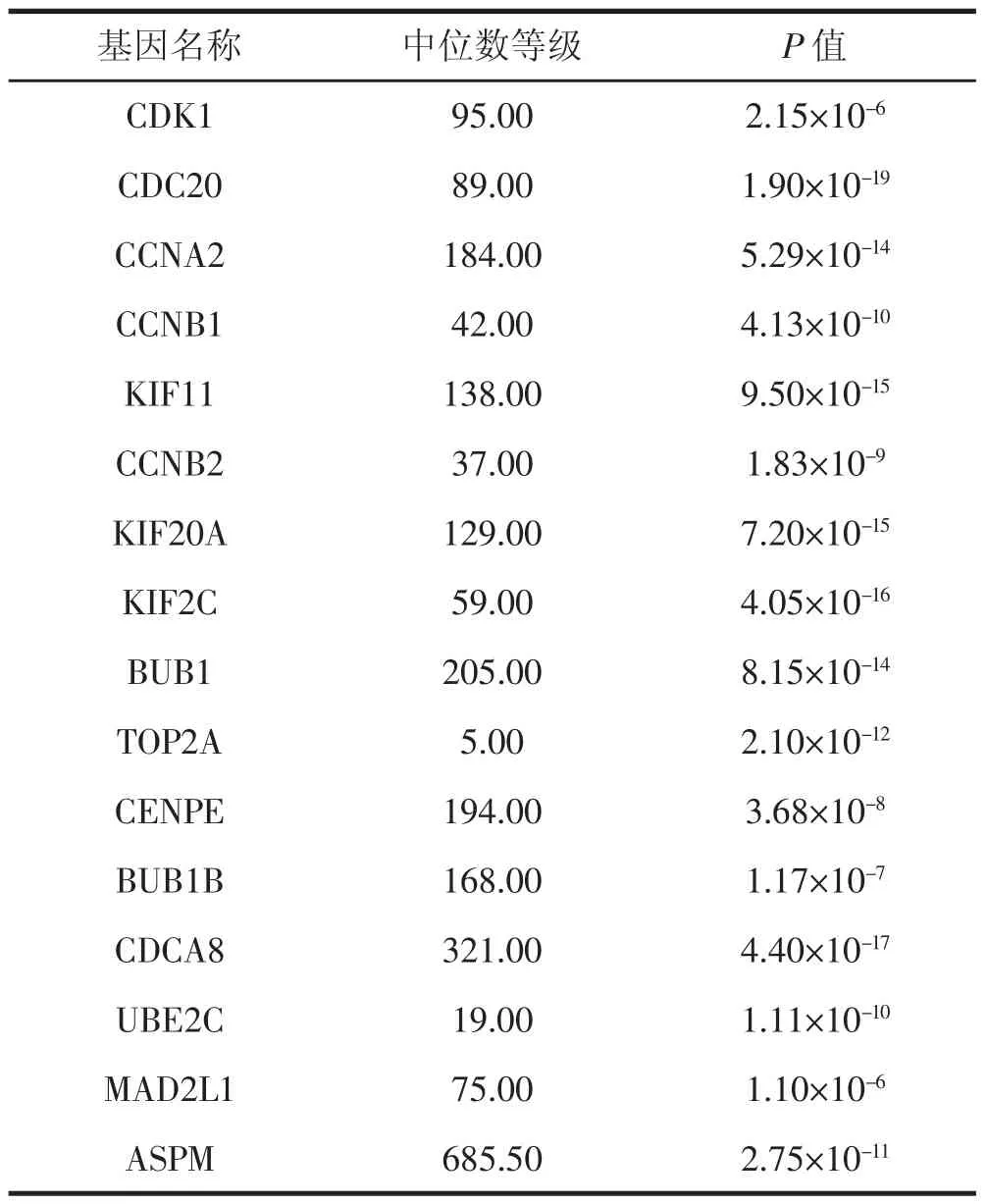
表3 Oncomine 数据库meta 分析结果
2.6 癌症与正常人hub 基因相关的生存分析
总生存时间是指从随机化开始至因任何原因引起死亡的时间,总生存期的延长可以体现确切的临床获益,是抗肿瘤药物最可靠的疗效评价指标。表4 生存分析结果将病人依据表达水平的中位数分为高表达组和低表达组,对得到的hub 基因进行生存分析以探究基因表达量与总生存时间之间的相关性。所有hub 基因的高表达都与肺腺癌患者的总体生存时间缩短相关。这提示了这些hub 基因在不同程度上与肺腺癌的病理和生理有关,可以作为监测肺腺癌预后的潜在生物标志物以判断肺腺癌的严重程度、预测患者的生存时间,或是作为治疗靶点。
3 结论
1)共筛选出肺腺癌相关16 个hub 基因。从高通量GEO 数据库中获得基因表达谱序列号,并使用R中的“limma”包对DEGs 进行鉴定,筛选发现1433 个DEGs(上调523 个,下调910 个)。为确定差异基因所涉及的生物学过程及信号通路,我们进行了GO 富集分析和基因组的KEGG 富集分析,发现DEGs 主要与细胞外基质组织、细胞外结构组织和有丝分裂的细胞核分裂有关,主要富集在补体和凝血级联、ECM受体相互作用和黏着作用的通路上。采用PPI 分析得到了由716 个DEGs 网络节点和3759 条边构成的PPI 网络,并在其中初步筛选出20 个hub 基因。通过WGCNA分析从DEGs 中得到了18 个模块,对hub 基因进行鉴定,筛选出17 个hub 基因。使用Oncomine 数据库对hub 基因在肺腺癌患者与正常人的表达情况进行Meta 分析,共鉴定出16 个hub 基因与肺腺癌相关。16个hub 基因分别为BUB1B、CDK1、CDC20、CCNA2、CCNB1、KIF11、CCNB2、KIF20A、KIF2C、BUB1、TOP2A、CENPE、CDCA8、UBE2C、MAD2L1、ASPM。
2)16 个hub 基因通常在肺腺癌患者中过表达。分析表明肺腺癌表型16 个hub 基因表达高于正常表型,据KEGG 分析,这些hub 基因主要与细胞周期、细胞衰老和p53 信号通路有关。GO 分析也证明它们显著参与了细胞核分裂,细胞器分裂和染色体分离过程。根据KEGG 分析和GO 分析的结果,这些基因与染色体的变化有关,可能在肿瘤的发生和发展中起到重要作用。
3)hub 基因高表达的组总体生存时间低于低表达的组。癌症与正常人hub 基因相关的生存分析发现,在肺腺癌患者中,这些hub 基因高表达的组总体生存时间下降。因此,这16 个hub 基因可能与肺腺癌的病理和生理密切相关,也可能是潜在的生物标志物。
4 展望
2 份关于LUAD 的数据集在筛选后一共发现1433 个差异表达基因,通过GO 和KEGG 等注释工作发现其DEGs 功能板块联系紧密,共涉及53 条生物学通路。这53 条通路和16 个hub 基因为LUAD 发生和发展提供了更详细的分子机制,有望作为潜在的生物标志物和治疗靶点。虽然肺腺癌的治疗在近年有所改善,但它仍然是最具攻击性和致命性的肿瘤类型之一[26]。肺腺癌分子机制的识别对肺腺癌的诊断、治疗和预后至关重要。微阵列基因表达谱被广泛用于筛选参与肿瘤发生的DEGs,为临床提供有价值的信息[27]。
癌症基因普查是一项正在进行的工作,目的是对那些包含与癌症有因果关系的突变的基因进行分类,并解释这些基因的功能异常如何导致癌症[28]。在16个hub 基因中,BUB1B 编码参与纺锤体检查点功能的激酶,并在细胞分裂过程中控制染色体分离[29];CDK1编码的蛋白质对于真核细胞G1/S 和G2/M 期的过渡至关重要[30];CDC20 表达的抑制可降低细胞增殖并诱导G2/M 细胞周期阻滞[31];CCNA2 通过整合素αVβ3信号通路促进肺癌细胞的侵袭和迁移[32];CCNB1 编码一种有丝分裂调控蛋白[33];KIF11 在纺锤体功能中起关键作用[34];CCNB2 编码细胞周期蛋白,表达下调能抑制肺腺癌的发生[35];KIF20A 敲除的可改变肺癌细胞表型并调控JNK 通路[36];KIF2C 编码微管解聚和染色体分离的调节因子[37];BUB1 编码丝氨酸/苏氨酸蛋白激酶与激活纺锤体启动子中发挥作用[38];TOP2A是公认的致癌基因;CENPE 可以促进肺腺癌细胞的增殖[39];CDCA8 的表达能刺激的乳腺癌细胞生长和存活[40];UBE2C 编码E2 泛素结合蛋白参与了细胞癌变[41];MAD2L1 是有丝分裂纺锤体装配检查点的1 个组件[42];ASPM 通过调节CDK4 参与肺鳞癌的发生发展[43]。这16 个hub 基因与细胞周期、细胞核分裂、染色体分离等细胞分裂活动相关,但它们能否作为肺腺癌预测、诊断、个体化预防、治疗和预后的生物标志物有待进一步实验验证。
猜你喜欢
动物医学进展(2024年4期)2024-04-10 01:50:04
家庭医学(下半月)(2020年1期)2020-05-11 02:05:32
心电与循环(2020年1期)2020-02-27 07:48:24
中成药(2018年7期)2018-08-04 06:04:18
海峡姐妹(2018年7期)2018-07-27 02:30:36
特别健康(2018年4期)2018-07-03 00:38:08
特别健康(2018年2期)2018-06-29 06:13:42
西南军医(2016年3期)2016-01-23 02:17:47
中国当代医药(2015年17期)2015-03-01 02:03:36
湖北农业科学(2014年3期)2014-07-21 10:25:00
