
CRISPR/Cas 系统在基因修饰植物及其产品检测应用中的原理和进展
2023-12-13 02:12:22王渭霞朱廷恒赖凤香万品俊魏琪傅强
中国稻米 2023年6期
王渭霞 朱廷恒 赖凤香 万品俊 魏琪 傅强
(1 中国水稻研究所/水稻生物育种全国重点实验室,杭州 310006;2 浙江工业大学 生物工程学院,杭州 310014;第一作者:wangweixia@caas.cn;*通信作者:thzhu@zjut.edu.cn;fuqiang@caas.cn)
成簇规律间隔短回文重复序列(clustered regularly interspaced shorted palindromic repeats, CRISPR)及其相关蛋白(CRISPR-associated protein, Cas 蛋白) 系统CRISPR/Cas 是古细菌等微生物在长期进化过程中形成的一种抵御外来DNA 入侵的保护机制[1-2],通过该系统将初次入侵生物的部分保守DNA 短序列剪切后作为间隔序列整合到重复序列之间形成记忆库。整合的外源生物DNA 转录的crRNA(CRISPR RNA)可精准识别再次入侵生物DNA,并启动该系统的核酸切割功能,从而保护自己不被侵染裂解死亡[3-4]。基于CRISPR系统精准识别和切割基因组的功能发展而成的基因编辑技术像一把“魔剪”,不仅能“剪切”基因,还能用于“修补”基因。该技术在生命科学和技术领域显示出强大的生命力,可以对靶标基因进行敲除、插入和碱基定点替换等精准编辑,在物种基因组修饰、基因工程等方面显示出广阔的应用前景。在农业领域可用来提高作物产量、增加抗性、改良品质以及创制雄性不育和固定杂种优势[5-6]。
CRISPR/Cas 系统的开发应用越来越广泛,除了crRNA 引导的靶标DNA 区精准编辑,部分Cas 蛋白还会通过Cas 蛋白/crRNA/靶DNA 三元复合物启动非特异性降解单链DNA(ssDNA)/单链RNA(ssRNA),也被称为反式切割。基于此,CRISPR/Cas 系统也被广泛应用于快速分子检测,包括病原微生物、单核苷酸突变等检测领域,展现出特异性高、灵敏度高、无需特殊仪器、快速准确等突出优势[7-9]。本文综述了CRISPR 系统在转基因以及基因编辑产品检测中的应用进展。
1 CRISPR/Cas 系统介绍
1987 年,ISHINO 等[10]在对K12 大肠杆菌中的碱性磷酸酶基因测序时发现,其附近有许多串联间隔的重复序列,2002 年JANSEN 等[11]将其命名为CRISPR 序列。进一步发现CRISPR 的间隔序列与噬菌体的某些序列存在着高度同源性[1],随后证实细菌可利用CRISPR 系统抵御噬菌体入侵和外源质粒的转移[12-13]。CRISPR 的保守重复序列长度为25~50 bp,间隔片段长度为26~72 bp,二者间隔串联而成[14],这些串联重复序列源于不同时间、不同噬菌体的感染整合,存在于40%的细菌和90%的古细菌中[12]。自2011 年起,CRISPR/Cas 系统介导的免疫机理不断被揭示,CRISPR/Cas 系统由CRISPR 基因座、Cas 蛋白和tracrRNA(trans-activating RNA)三部分组成。CRISPR 的前导序列转录为前体crRNA(pre-CRISPR-derived RNA),pre-crRNA经RNA 内切酶切割生成含有与噬菌体靶标序列匹配的成熟CRISPR RNA ( crRNA),crRNA 通过碱基配对与tracrRNA 结合形成tracrRNA/crRNA 复合物,此复合物引导Cas 蛋白实现对靶标序列的识别和切割[15]。Cas 蛋白对靶标序列的识别必须依赖于间隔序列毗邻基序( Protospacer adjacent motif,PAM)。tracrRNA 的功能主要是连接crRNA 和Cas 蛋白。后来通过人工设计将crRNA 和tracrRNA 改造合并成一条具有引导作用的sgRNA(single guide RNA),可同样实现引导Cas 蛋白对目标DNA 的定点切割[16]。
CRISPR/Cas 系统根据其组成和功能可分为两大类群:第一类由多个Cas 组成的效应复合体行使功能;第二类则由单个多结构域的Cas 组成,包括Ⅱ型的Cas9、Ⅴ型的Cas12 和Ⅵ型的Cas13[17]。第二类系统简单,蛋白分子量小,操作方便,是目前主要应用的基因编辑系统。其中,通过融合胞嘧啶脱氨酶和尿嘧啶糖基化酶抑制剂的Cas 蛋白可高效地对靶向位点内的特定碱基进行替换,进一步提高了定点基因精准编辑的功能[18-19]。
2 CRISPR/Cas 在分子检测中的应用
在Cas 蛋白切割靶标DNA 的同时,发现某些Cas蛋白会启动其附带“切割活性”,完成对其他非靶标DNA 或RNA 的任意切割。这一特性为检测样品是否含有某目的DNA/RNA 序列提供了可行性。例如,合成含特异性识别靶标序列(目标序列)的sgRNA 和切割非特异性序列(荧光报告核酸)的检测系统,一旦sgRNA 检测到目的序列,系统将启动,荧光报告核酸也会被降解,从而释放出荧光信号。截至目前,CRISPR/Cas系统里有三类单一效应蛋白(Cas12、Cas13 和Cas14)被报道具有非特异性切割核酸特性,可用于分子诊断检测[20-24]。基于不同Cas 的核酸切割功能,开发出了SHERLOCK (Cas13a/RNA)、DETECTR [Cas12a/dsDNA(双链DNA)或Cas14a/ssDNA]等核酸检测系统(图1),并成功应用于SARS-CoV-2、寨卡病毒、埃博拉病毒、HPV 和结核分枝杆菌等的检测[9,25]。

图1 三种Cas 蛋白附属切割活性进行分子诊断的示意图[9]
2.1 基于Cas13a 的分子检测
Cas13a 属于第二类中VI 型CRISPR-Cas 蛋白,可在gRNA 的引导下靶向单链RNA,激活单链RNA 酶活性,并启动非特异性切割RNA 特性[26]。DOUDNA 团队设计了一端带有荧光素另一端带有淬灭集团的荧光报告RNA,Cas13a 在匹配到靶标RNA 后即可非特异性切割环境中的荧光报告RNA,释放荧光素并发出荧光,从而实现目标RNA 检测。采用该技术成功检测了噬菌体RNA,灵敏度达0.01 nM[20]。基于此,重组酶聚合酶扩增(recombinase polymerase amplificatin,RPA)相结合设计出SHERLOCK(specific high-sensitivity enzymatic reporter unlocking)技术,使得样本中低浓度的靶标分子DNA 首先通过RPA 扩增后转录或RNA 分子通过逆转录重组酶聚合酶扩增(RT-RPA)得到指数级扩增,再采用Cas13a-gRNA 与荧光报告RNA 检测,大幅提高了检测灵敏度。可在2 h 内直接从患者样本(如血清、尿液和唾液)中以低至每1 μL 1 个拷贝的浓度直接检测寨卡病毒(ZIKV)和登革热病毒(DENV)[27]。在升级版的SHERLOCKv2 中,使用4 种不同的Cas13和Cas12 酶的组合,在单个反应中可检测到4 种靶标核酸序列,并通过使用CRISPR 相关酶Csm6 提高Cas13 活性,使检测灵敏度提高了约3.5 倍[28]。
2.2 基于Cas12a 的分子检测
Cas12a 是第二类V 型CRISPR-Cas 蛋白,含有RuvC 催化结构域可识别富含T 的靶向序列,在sgRNA的引导下特异性切割双链DNA(dsDNA)或单链DNA(ssDNA)并产生附属的ssDNA 酶活性[21,23]。基于此,通过将CRISPR/Cas12a 系统与RPA 相结合,开发了一种新的DNA 内切酶靶向CRISPR 反式报告器DETECTR(DNA endonuclease-targeted CRISPR trans reporter)用于核酸检测(图1)。该技术被成功应用于HPV 病毒的分型检测[21],DNA 病毒pseudorabies virus (PRV) 和RNA 病毒Japanese encephalitis virus (JEV)的检测[23]。与SHERLOCK 相比,该技术体系减少了从扩增后的DNA转录到RNA 这一步,其荧光报告分子为DNA 探针,易于保存。随着类似于Cas12a 活性的其他Cas 蛋白如Cas12b 等的发现,基于Cas12 的核酸检测系统得到了一系列改进,集成了多种扩增方法(包括PCR、RPA、LAMP)和单一读数形式(例如荧光检测器、裸眼观察和侧向流动分析),使该方法更灵敏、准确、便携且更易于使用[25,29]。
2.3 基于Cas14a 的分子检测
古细菌中发现一种具有400~700 个氨基酸组成的属于第二类系统Ⅴ型的Cas14 蛋白,具有典型的RuvC核酸酶结构域,可靶向ssDNA,在激活后剪切靶ssDNA和非特异性地剪切环境中的ssDNA[22]。Cas14 对靶标序列没有前间区序列邻近基序的限制,所以可以靶向任何序列,但对识别序列中间的碱基的要求很严格,有1个错配就不能结合,特异性能达到单碱基。但由于Cas14-sgRNA 只识别ssDNA,因此在检测应用中需要对靶序列扩增的引物进行修饰,如使用含硫代磷酸(phosphorothioate oligonucleotide,PT)的引物扩增DNA底物,以保护一条链不被核酸外切酶降解。扩增产物加入T7 核酸外切酶后,带有未修饰引物的链被降解,留下一条可被Cas14a 检测的ssDNA 底物。利用该系统成功区别了与人类眼睛颜色有关的HERC2基因SNP(图2),首先通过设计一条与蓝色眼睛的目标序列严格互补sgRNA,与sgRNA 互补链的引物含有PT,对唾液样本中HERC2基因进行扩增。扩增产物经T7 核酸外切酶酶切后,得到的蓝色眼睛ssDNA 与sgRNA 互补并启动Cas14a 的ssDNA 切割活性,随后启动其非特异切割活性,使荧光报告探针释放出荧光得到检测,而仅有1个碱基之差的(G-T)的褐色眼睛的ssDNA 得不到检测。而Cas12 系统则无法区别这种单碱基突变的样本。因此,通过将Cas14 的非特异性ssDNase 切割活性与等温扩增法相结合(DETECTR-Cas14),可用于高保真度DNA 单核苷酸多态性基因分型,可能在传染性和非传染性疾病的CRISPR 诊断领域获得指数级扩展[30]。
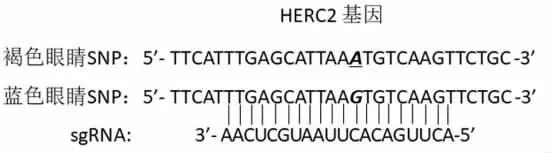
图2 利用Cas14 系统检测人类HERC2 基因单碱基突变的示意图
3 基于CRISPR/Cas 系统的转基因检测
根据检测对象不同,基于核酸的植物转基因检测技术包括对常用的转基因元件启动子(CaMV35S、FMV35S)、终止子(NOS、E9’3、T-CaMV35S)、选择标记基因(NPTII、HPT)及目的基因(Bt、Bar、Pat、EPSPS)筛选检测,对载体构建的构建特异性检测,以及对插入受体基因组的边界序列的转化体特异性检测。检测通常采用定性PCR、荧光定量PCR 和数字PCR 等方法。常规PCR 需要在扩增仪上经过变性、退火、延伸三个步骤来完成扩增。环等温扩增技术(loop-mediated isothermal amplification, LAMP)和RPA 反应最适温度分别为65 ℃和37 ℃,不需要复杂的温控设备,可以真正实现便携式快速核酸检测,从而替代PCR 检测技术[31-32]。CRISPR/Cas 检测结合LAMP 或RPA 技术有望实现转基因产品的现场快速检测。其中,Cas12 检测靶标为双链DNA 分子,与转基因靶标的双链DNA 一致,因此,Cas12 最有望应用于转基因产品的检测。
3.1 基于Cas12a 的转Cry1C 基因的水稻的检测
结合RPA 技术,通过合成sgRNAs 引导Cas12a 蛋白结合到RPA 扩增产物和单链DNA 报告探针(5-FAM-TTATT-Quencher-3),对转Cry1C基因水稻实现高效检测[33]。设计了4 种与Cry1C不同位置互补的sgRNA(1-4)引导Cas12a,检测了对RPA 扩增产物和报告探针的切割活性。结果发现,Cas12a 的靶标DNA切割活性独立于单链DNA 报告探针切割活性,检测能力主要依赖于gRNA,其中sgRNA1 和sgRNA3 对靶标DNA 的切割活性可达100%,sgRNA1 对DNA 报告探针的切割活性显著高于其他sgRNA。
该体系只需要合成长度为30~35 nt 的RPA 扩增用引物,基因组DNA 与扩增酶在缓冲液中37 ℃温浴10 min 获得RPA 扩增产物,随后添加1 μM Cas12a、100 ng sgRNA 和1 μL 单链DNA 报告探针,37 ℃温浴10~60 min。反应产物通过胶体金试纸条就可以在5~10 min 内获得检测结果(图3)。该技术体系要点:RPA扩增产物需含有Cas12a 蛋白切割识别的PAM 序列TTTG;其次紧随PAM 序列后的sgRNA 序列与目标序列的一致性是检测特异性的关键,错配1 个碱基都会影响Cas12a 蛋白的切割活性。sgRNA 序列包含有引导序列和保守的茎环序列。以sgRNA1 为例:RPA 扩增引物分别设计在Cry1C基因序列(GenBank 登录号:HM107006.1)的156~185 bp 和506~537 bp 处,扩增产物位于156~537 处,长度328 bp。sgRNA1 则设计在227~250 bp处,序列为5’-TCGTTCAGATCGAACAGCTCATCA-3’,上游PAM 序列为223-TTTC-226。首先合成寡核苷酸序列sgDNA:5’-tgatgagctgttcgatctgaacgaATCTACAACAGTAGAAATTCCCTATAGTGAGTCGTATTAATTTC -3’,其中小写字母为靶标序列(227~250 bp)的互补序列,大写字母为T7 启动子互补序列,大写斜体为形成sgRNA茎环结构的核心序列,该序列通过与T7 启动子序列退火合成双链后即可利用体外转录试剂盒转录获得sgRNA1(图4)。
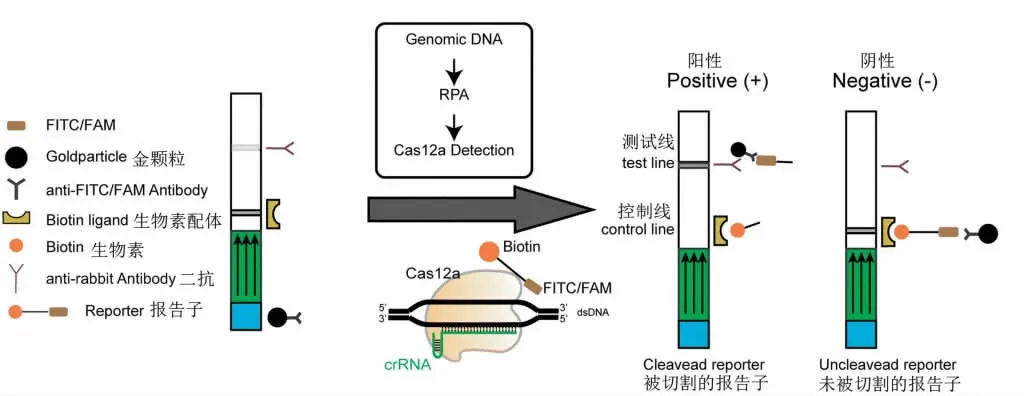
图3 基于Cas12a 的侧向流动检测示意图[33]
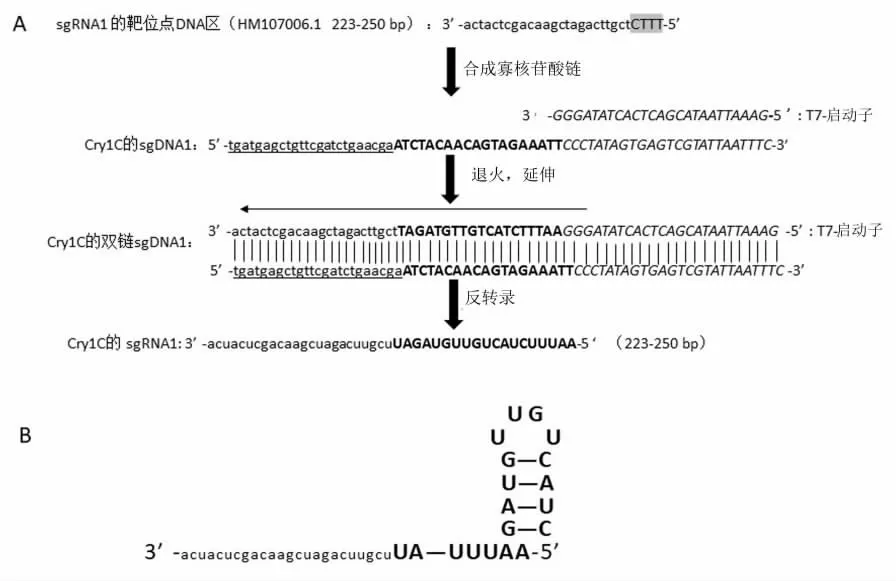
图4 Cry1Ac 的sgRNA1 序列合成示意图(A)和二级结构(B)
3.2 基于Cas12a 的CaMV35S 启动子的检测
结合LAMP,对转基因植物的外源启动子CaMV35S(GenBank 登录号GU734659.1)的644~934 bp处进行扩增。扩增片段中需含有可以激活Cas12a 活性的PAM 序列即富含T 的TTTN,单链DNA 报告探针为5’6-FAM-TTATT-3’BHQ1 和可引导Cas12a 蛋白靶向LAMP 扩增产物中CaMV35S扩增子的sgRNA。sgRNA 包括可以形成茎环结构的核心序列UAAUUU CUACUAAGUGUAGAU 和目标序列CUUUAUCGCAA UGAUGGCAUU。目标序列DNA 位于GU734659.1 717~737 bp 处,序列为CTTTATCGCAATGATGGCATT,上游为PAM 序列TTTC。在sgRNA 中只是“T”被“U”取代(图5)。在37 ℃下CRISPR/Cas12a 系统与扩增产物温浴5 min 就可在紫外光下肉眼快速识别检测结果。所建立的方法能显著区分特异性扩增和非特异性扩增。检测限可达转基因含量0.05%[34]。将LAMP 反应和Cas12a 检测试剂混合在1 个反应管中可有效防止源头污染,无需额外试剂,使操作过程更加快捷方便。

图5 CaMV35S 的sgRNA 序列二级结构和CaMV35S 扩增子的对应靶标DNA 序列示意图
4 展望
转基因检测中,开发低成本、准确、高效和快速的检测方法非常重要。相较于PCR 为基础的检测技术,CRISPR/Cas 系统显示出其优越性,有助于实现多种基因修饰产品的高灵敏度、高精度检测,包括现场快速检测。
CRISPR/Cas 检测系统显著拓宽了可检测对象核酸种类。充分利用Cas 蛋白的附属切割活性多样性功能,可对不同种类和构象的靶标核酸进行精准检测。Cas12 擅长识别双链DNA,Cas13 擅长识别单链RNA,而Cas14 则擅长识别单链DNA。这对提高不同基因修饰产品及其不同核酸样品的准确率提供技术保障。
CRISPR/Cas 检测技术在基因修饰产品的检测应用中才刚刚起步。通常,针对不同转基因产品,可开发一套常用元件的基因编辑检测技术以及身份特异性快速检测,应用于现场快速检测对转基因产品的监管有重要意义。目前已经开发出的针对Cry1C和CaMV35s启动子的CRISPR/Cas 检测技术为转基因植物的筛选检测开启了先河。针对每一个市场化转基因产品的身份特异性快速检测可为育种者知识产权保护以及杜绝假种行为提供支撑,该技术的关键是合成特异性强、可诱导强切割活性的sgRNA。因此,针对特定检测的靶标序列对比研究获得灵敏度高、特异性高的sgRNA 是实现不同转基因植物快速现场检测的先决条件。
相比于转基因技术即将异源物种DNA 导入到目标物种内并随机整合到目标物种染色体上的技术,利用CRISPR/Cas 的基因编辑技术是对物种自身靶基因实施的定点修饰,在此过程中会有载体序列等外源DNA 序列残留。与传统转基因植物被插入大片段外源基因序列的情况不同,基因组编辑技术在改造的受体生物体内留下的痕迹较少,已有的转基因产品检测方法不能完全适用,给基因组编辑植物及其产品的检测带来新的挑战[35]。对于含有外源短片段DNA 的基因编辑产品可借助于第二代测序技术和FED(foreign element detection)网络大数据库实现外源DNA 成分的检测识别[36]。FED 已经内置了24 种植物和13 种动物的参考基因组信息,整理归类了美国国立生物技术信息中心(National Center for Biotechnology Information)和addgene(https://www.addgene.org)等生物数据库收集的各国科研人员递交的不同物种的各类载体序列信息,并依此建立了外源成分数据库。而对于无外源DNA 引入的基因编辑修饰产品,尤其是单核苷酸改变的情况,主要依赖DNA 测序、限制性内切酶分析法、T7 核酸内切酶分析法、单链构象多态性分析方法、高分辨率熔解曲线分析法和荧光探针PCR 检测等极为复杂的检测手段[37-38]。这些检测方法耗时长、需要昂贵的仪器设备,且检测的准确率也有待提高。基于基因编辑系统的sgRNA 与靶序列的准确识别功能有望实现对单核苷酸突变的基因编辑产品的准确识别和检测。
猜你喜欢
学与玩(2022年10期)2022-11-23 08:32:00
中华诗词(2022年9期)2022-07-29 08:33:50
中国慈善家(2022年3期)2022-06-14 22:21:55
今日农业(2022年3期)2022-06-05 07:12:08
快乐语文(2021年34期)2022-01-18 06:04:14
军民两用技术与产品(2021年10期)2021-03-16 06:05:10
中国(俄文)(2020年8期)2020-11-23 03:37:13
世界农药(2019年3期)2019-09-10 07:04:10
光学精密工程(2016年4期)2016-11-07 09:04:48
创新科技(2015年1期)2015-12-24 06:23:21
