
木栓脂加固处理对饱水古木吸湿性及尺寸稳定性的影响
2023-12-05 11:11:34吕偲琪阚玉娜王军翟胜丞
林业工程学报 2023年6期
吕偲琪,阚玉娜,王军,翟胜丞,3*
(1. 南京林业大学材料科学与工程学院,南京 210037;2. 南京市考古研究院,南京 210004;3. 南京林业大学林业资源高效加工利用协同创新中心,南京 210037)
在长期的埋藏过程中,受到环境中厌氧细菌、软腐真菌等生物因素及酸、碱、盐等非生物因素的影响,考古木材主要化学组分如纤维素、半纤维素及木质素等均发生不同程度的降解。根据保存状态的差异,可将考古木材分为两种类型:干燥环境出土的考古木材和潮湿环境出土的饱水考古木材[1]。干燥环境出土的考古木材在降解过程中容易发生变形、干裂、扭曲及严重的坍塌变形,保存下来的文物较少。饱水考古木材在长期地下水环境中主要遭受了细菌等微生物的攻击,虽然细胞壁发生降解,其外观形貌却大都保持完整,但是出土后的饱水考古木材可能由于保护不当发生二次破坏,自然脱水后发生不可逆的收缩现象,古木的尺寸稳定性也因此下降。因此需要对出土饱水古木进行及时且适当的加固保护处理,以保持其原有的形态和外观,实现饱水木质文物的长期保存和馆藏陈列。饱水木质文物常用的保存方法有饱水保存和脱水保护,更常用的方法是对饱水木质文物进行脱水置换加固,从而实现木质文物的合理和长期保存。19世纪以来,保护科学领域的专家已经研究了多种对各类木制工艺品进行处理的方法,目前聚乙二醇(polyethylene glycol, PEG)、糖类、天然树脂、石蜡、挥发性溶剂置换等常用于饱水木质文物的加固保护,处理手段简单有效,但是长期保护过程中木材表面颜色加深,纹理模糊且性能出现了下降的情况[2-4]。质量增加率和尺寸抗收缩率(anti-shrink efficiency, ASE)是评估饱水古木加固效果的首要因素。Endo等[5]使用质量分数40%的鸭毛角蛋白溶液对糙叶树(Aphanantheaspera)、香樟(Cinnamomumcamphora)和日本常青橡树(Quercusacuta)等饱水考古阔叶材进行加固处理,研究发现加固古木的抗收缩率均超过60%,其中最大含水率为390%的日本常青橡树ASE值高达107.7%,尺寸稳定性提升。此外,吸湿性对木材尺寸稳定性有重要影响,木材吸湿性能的研究对饱水木质文物的加固与尺寸稳定性的长期保持有指导性意义[6-7]。
自然界中木栓脂主要存在于高等植物的外皮和块茎周皮中,在与工业相关的阔叶材如桦木、栎木中,木栓脂通常占无抽提物树皮质量的20%~50%[8]。木栓脂是栓皮栎(Quercusvariabilis)树皮细胞壁主要组成成分[9],化学界学者将木栓脂定义为脂肪族高分子物质[10]。木栓脂具有较好的耐腐性、耐热性、耐老化性及防水性,主要功能之一是对扩散有屏蔽的作用,水分无法通过细胞壁,使得栓皮栎树皮的化学性质十分稳定,木栓脂的优良性质使得栓皮栎树皮具有对液体的不渗透性以及对化学物质和细菌的阻隔性[11]。栓皮栎树皮中木栓脂质量分数为37%~50%,部分研究中木栓脂质量分数甚至高达62%[12-13]。双癸基二甲基氯化铵(C22H48ClN, didecyl dimethyl ammonium chloride, DDAC)表面活性剂是季铵盐的一种,是低毒、环保的烷基胺化合物,广泛应用于消毒剂、杀菌剂及木材防腐剂等领域[14-16]。本研究提取栓皮栎树皮中的木栓脂,采用双癸基二甲基氯化铵活性剂作为乳化剂制备水溶性木栓脂加固剂对饱水木质文物进行加固处理,系统研究了加固后考古木材质量增加率、尺寸稳定性变化,采用显微观察技术分析木栓脂加固剂在古木中的分布,结合湿化学法和傅里叶变换红外光谱技术获取加固古木细胞壁化学结构变化,探索木栓脂加固液对考古木材的加固机理。结合平衡含水率变化以及建立吸湿等温线Guggenheim-Anderson-de Boer(GAB)模型对加固古木的吸湿性能进行表征,评估木栓脂加固古木的效果,为饱水木质文物的保护提供科学依据。
1 材料与方法
1.1 试验材料及试剂
饱水考古木材(南京大阳沟宋代墓葬群出土),松科(Pinaceae)硬松(PinussubgenusDiploxylonspp.);栓皮栎树皮取自大自然阿豪软木公司;三氟乙酰胺、三甲基硅烷、双癸基二甲基氯化铵、硫酸、C2H3KO2、MgCl2、K2CO3、NaBr、NaCl和KCl均为化学纯,来自南京化学试剂有限公司。
1.2 木栓脂加固液的制备
参考Ferreira等[17]的提取方法进行木栓脂提取。将木栓脂提取液与DDAC、去离子水按质量比进行20%,25%,30%,35%和40%质量分数梯度的混合,搅拌均匀,配制木栓脂加固液,具体配制比例如表1所示。

表1 木栓脂加固液的配制Table 1 Composition of suberin-based consolidants
1.3 木栓脂加固液化学成分测定
利用气相色谱-质谱联用仪(gas chromatography-mass spectrometry, GC-MS,GC 7890A/MSD 5975C,美国Agilent公司)进行木栓脂化学成分的测试,选取VF-1701色谱柱(30 mm × 0.25 mm × 0.25 mm),载气为高纯氦气,流速为1 mL/min;柱温在40 ℃下保持2 min,以5 ℃/min速率升温至200 ℃,再以10 ℃/min速率升温至280 ℃,然后保持3 min。
1.4 加固考古木材质量增加率及抗收缩率测定
利用电子天平称量饱水木块的质量(m0),精确到0.001 g。参照GB/T 1933—2009《木材密度测定方法》测量饱水木块弦向、径向及轴向尺寸,利用排水法测量体积,之后将饱水木块按编号顺序放入离心管中,每个浓度设置3组平行样,分别加入配制好的木栓脂加固剂。将饱水试样置于真空干燥箱(DZF型,上海力辰邦西仪器科技有限公司)中真空浸渍1 h后取出,盖上密封盖浸渍7 d。其间每隔48 h测量1次古木的质量、弦向尺寸、径向尺寸、轴向尺寸以及体积,当试样质量变化小于0.1%时,渗透完成。利用真空冷冻干燥机(FD-1G-50,上海贺帆仪器有限公司)对加固古木与未处理饱水古木冷冻干燥24 h完成古木的脱水定型。
根据加固前后古木质量变化计算木材的质量增加率,计算公式如下:

(1)
式中:m2为加固古木冷冻干燥后的质量,g;mc为未处理古木冷冻干燥后的质量,g。
通过加固前后干燥试样的体积变化,即润胀率,判断细胞壁的润胀程度,计算公式如下:

(2)
式中:V2为加固古木冷冻干燥后的体积,mm3;Vc为未处理古木冷冻干燥后的体积,mm3。
作为衡量木材尺寸稳定性的标准,木材抗收缩率计算公式如下:

(3)
式中,βc、β2分别为未处理古木、加固古木冷冻干燥后的尺寸收缩率,参考GB/T 1932—2009《木材干缩性测定方法》,可由式(4)、(5)计算得出:
(4)
(5)
式中:L0为饱水木块轴向尺寸,mm;Lc为未处理古木冷冻干燥后的轴向尺寸,mm;L2为加固古木冷冻干燥后的轴向尺寸,mm。
1.5 加固考古木材色度的测定
根据国际照明委员会颁布的CIE标准色度系统使用色彩色度仪(KONICA CR-5型,日本)对未处理古木与加固古木的明度(L*)和色品指数(a*、b*)进行测定,在样品表面选取3个点,每点测量3次,取平均值。其中:L*为明度,0表示全黑,100表示全白;a*为红绿色品指数,正值表示偏红(数值越大,颜色越红),负值表示偏绿;b*为黄蓝色品指数,正值表示偏黄(数值越大,颜色越黄),负值表示偏蓝。总体综合色差(ΔE*)计算公式[18]如下:
(6)
式中,ΔL*、Δa*、Δb*分别表示不同试样的明度、红绿色品指数和黄蓝色品指数与标准颜色对照参数的差值。
1.6 加固考古木材中加固剂分布及渗透能力测试
1.6.1 扫描电镜观察
利用YAMATO滑走式切片机制备古木横切面切片(厚度为100 μm)用于微观形貌观察。对样品表面喷镀铂金以提高导电性能,并通过扫描电子显微镜(Phenom XL,荷兰Phenom公司)进行数据获取,电流不可超过10 mA,加速电压为10 kV。
1.6.2 透射电镜观察
利用超薄切片机(Leica EM UC7,德国Leica)对树脂包埋后的样品进行超薄切片,切片厚度70 nm。采用1% KMnO4染液对超薄切片进行染色,染色时间为10 min。利用透射电子显微镜(JEM-1400,日本电子株式会社)对超薄切片进行超微构造的观察。
1.6.3 傅里叶变换红外光谱测试
将木块磨成粒径为250~380 μm(40~60目)的木粉,烘至绝干备用。采用红外光谱仪(Frontier,美国PerkinElmer公司)对试样进行傅里叶变换红外(Fourier transform infrared, FT-IR)光谱测试,扫描范围为4 000~500 cm-1,扫描次数为32次。随后用PerkinElmer Spectrum软件(美国PerkinElmer公司)进行数据处理,以1 030 cm-1附近吸收峰为基准归一化。
1.7 加固考古木材主要化学组分测试
取粒径250~380 μm(40~60目)木粉,参考GB/T 2677.6—1994《造纸原料有机溶剂抽出物含量的测定》对木粉进行索氏抽提以去除木材中的抽提物。饱水考古木材与现代健康材中的纤维素、半纤维素及木质素的相对含量的测定按照美国能源部可再生能源实验室的方法进行测试[19]。准确称取0.3 g的绝干脱脂木粉样品放入水解瓶中,加入3 mL的72%硫酸,30 ℃水浴锅中震荡1 h,加入84 mL去离子水,置于121 ℃高温高压灭菌锅水解1 h,使用G2玻璃砂芯过滤。木质素分为酸溶木质素和酸不溶木质素,过滤后的滤渣用于测试酸不溶木质素,计算公式如下:

(7)
式中:G1为灼烧后的灰分和玻璃滤器的质量,g;G2为过滤后残渣和玻璃滤器的绝干质量,g;W为原料绝干质量,g。
使用紫外分光光度计(UV-1800,日本SHIMADZU)在205 nm波长下测试酸解液的吸光度,计算酸溶木质素的质量分数,计算公式如下:

(8)
式中:D为稀释倍数;A为紫外吸收值,205;V为酸解液体积,mL;110为吸收系数;W为原料绝干质量,g。
原料中纤维素和半纤维素含量由酸解液中的单糖含量计算得到,计算公式如下:

(9)

(10)
式中:V为酸解液体积,mL;Cg、Cx和Ca分别为酸解液中的葡聚糖、木糖和阿拉伯糖浓度,g/L;W为原料绝干质量,g。
单糖含量通过高效液相色谱仪(Agilent 1260 series,美国Agilent公司)测定。
1.8 加固考古木材吸湿特性测试及模型拟合
将加固木块切成1 mm (径向) × 1 mm (弦向) × 3 mm (轴向)的长方体木条试件用于吸湿性能测试。分别配制C2H3KO2、MgCl2、K2CO3、NaBr、NaCl和KCl的饱和盐溶液,倒入干燥器内进行调湿处理。为保证盐溶液始终处于饱和状态,配置固液比为1∶3,静置12 h等待环境湿度达到设定值不再变化(即固、液、气三相平衡)。25 ℃时饱和盐溶液对应的相对湿度如表2所示。

表2 25 ℃饱和盐溶液对应的相对湿度Table 2 The relative humidity corresponding to the saturated salt solution at 25 ℃
将绝干试样置于离心管中,按顺序分别放入环境湿度为23%,33%,43%,58%,75%和84%的干燥器内静置48 h,每24 h称量1次质量,待到试样质量变化小于0.1%时,计算样品含水率。计算公式如下:

(11)
式中:m3为绝干样品质量,g;m4为吸湿后样品质量,g。
利用Guggenheim-Anderson-de Boer(GAB)模型分析加固古木的吸湿特性曲线,以及在等温吸湿过程中的平衡含水率、单分子层的饱和吸附量和比表面积。GAB模型是由Origin软件结合最小二乘法模拟获得,计算公式如下:
(12)
式中:Vw为单位质量吸附水体积,cm3/g;Vm为单层分子饱和吸附量,cm3/g;C为单层分子吸附水与多层分子吸附水之间自由焓(相较于标准化学势)之差相关的常数;K为多层分子吸附水与纯水自由焓之差相关的常数;φ为等温吸附过程相对湿度。
GAB模型单层分子吸附水所占的面积就是木材细胞壁吸附作用有效比表面积,计算公式如下:
(13)
式中:S为单位质量木材有效比表面积,m2/g;Vm为单层分子饱和吸附量,cm3/g;ρ为吸附水密度,g/cm3,取1 g/cm3(默认木材细胞壁内吸着水密度与自由水相同);σ为1个水分子占据面积,均值取0.114 nm2;18为水的摩尔质量数值;NA为阿伏伽德罗常数,取值 6.022 × 1023。
2 结果与分析
2.1 加固饱水考古木材质量增加率及尺寸
木栓脂加固考古木材的质量增加率、尺寸收缩率和抗收缩率如图1所示。从图1a可以看出,当木栓脂加固液质量分数为35%时,质量增加率达到最大值69.35%;同时加固后古木细胞壁发生润胀,35%木栓脂加固古木润胀率达到最大值38.87%。当木栓脂质量分数增加至40%时,加固古木的质量增加率没有进一步提升。该现象可能因为进入木材细胞内的木栓脂存在浓度极限值,在此极限之前木栓脂都能进入细胞,随着加固剂浓度的增加,质量增加率呈现上升的趋势;然而超过浓度极限后,细胞腔和孔隙中填充了一定的木栓脂,当浓度再升高时,木栓脂无法进入细胞内部,对古木的填充效果没有进一步的提升。图1c中未处理考古木材的弦向收缩率、径向收缩率和轴向收缩率分别为17.45%,5.65%和5.07%;35%木栓脂加固的古木弦向、径向和轴向的各向尺寸收缩率均达到最小值,分别为3.58%,2.38%和0.82%;同时,弦向、径向和轴向的ASE均达到最大,分别为81.59%,57.88% 和83.83%,古木的尺寸稳定性提升效果最佳。结果表明,加固后古木细胞壁发生润胀,尺寸收缩率减小,抗收缩率增大,尺寸稳定性提高。

a)质量增加率;b)润胀率;c)尺寸收缩率;d)抗收缩率。
结合前人研究[5,20-22]与本试验加固考古木材尺寸稳定性变化进行对比,结果如表3所示。与PEG、甲基三甲氧基硅烷、木质素纳米粒子和纤维素纳米晶体相比,木栓脂加固古木弦向抗收缩率较高,古木的抗收缩性较强,加固效果更优。

表3 不同加固剂对考古木材尺寸变化的影响Table 3 Effects of different consolidants on the dimension change of the archaeological wood
2.2 加固饱水古木颜色的变化
木栓脂加固前后考古木材外观形态和色泽的变化如图2所示。图2a显示,饱水古木由于干燥过程中水分的流失发生了收缩变形。冷冻干燥后的考古木材(图2b)表面颜色暗淡,没有光泽。图2c是用质量分数35%木栓脂加固后冷冻干燥状态的古木,与饱水古木相比,加固古木的外观形态和纹理基本不变,形态几乎与饱水状态时相同,表面颜色较干燥状态下未处理古木略深,偏暗。结合饱水考古木材颜色差值变化可知(表4),与未处理古木相比,不同浓度木栓脂加固古木的明度差值(ΔL*)呈现下降趋势,下降了0.78~3.37;而红绿色品指数差值(Δa*)和黄蓝色品指数差值(Δb*)总体上均呈现增长趋势,总色差值(ΔE*)降低了0.40~1.71。结果表明,与未处理古木相比木栓脂加固古木的明度有所下降,表面略微变暗。

a)饱水古木;b)冷冻干燥古木;c)冷冻干燥加固古木;d)不同状态古木形态变化的模式图。注:图d中黑色线框表示饱水古木的外观形态,红色虚线框表示干缩变形古木的形态,蓝色线框表示饱水古木加固处理后冷冻干燥状态下的外观形态,标尺为2 mm。

表4 加固前后考古木材颜色差值变化Table 4 Changes of color difference of archaeological wood before and after reinforcement
2.3 加固剂的渗透能力及分布
未处理古木与35%质量分数木栓脂加固古木的扫描电镜图像如图3所示。图3a显示,未经过任何处理的古木次生壁与复合胞间层剥离,紧致的细胞壁结构变得蓬松,次生壁上出现蜂窝状孔隙,表明细胞壁主要化学组分已发生较大程度的降解。图3b~d为加固古木横切面微观形貌。加固古木的管胞腔中有大量木栓脂分布,加固剂在管胞腔内部分填充(图3b)或完全填充(图3c),但是仍有部分管胞腔未被填充,可能是由于加固剂的分散不均匀。与未处理考古木材相比,部分细胞壁层由于加固剂的沉积被薄膜包覆(图3d),加固处理后的古木细胞壁孔隙减少,壁层表面光滑平整;径切面管胞上的纹孔口也被加固剂填充,纹孔轮廓清晰可见,如图3f中箭头所示。木栓脂的沉积填充了细胞壁孔隙,降低孔隙率,提高了古木的尺寸稳定性。图4是未处理古木和加固古木的透射电镜图像,经1% KMnO4染色剂染色后,加固古木次生壁S2层中存在星散状的黑色物质区域,如图4b箭头所示;复合胞间层与细胞角隅处也观察到部分区域颜色较深,电子密集程度相对较高,如图4c~d中箭头所示。该现象可能是因为木栓脂加固剂进入细胞壁,不均匀地沉积在古木细胞壁次生壁S2层、复合胞间层以及细胞角隅处,导致木栓脂沉积之处颜色较深,电子相对密集。

a)未处理古木横切面;b~d)加固古木横切面;e)未处理古木径切面;f)加固古木径切面。

a)未处理古木;b~d)加固古木。S为细胞壁次生壁(S1、S2、S3),CML为复合胞间层。
2.4 加固饱水考古木材化学组分
2.4.1 主要化学组分相对含量
未处理古木与35%木栓脂加固古木主要化学组分相对含量如表5所示。加固考古木材的纤维素相对含量较未处理古木减少9.64%,木栓脂处理降低了古木纤维素的含量,但是木质素相对含量提升8.1%。

表5 未处理考古木材与加固考古木材的主要化学组分Table 5 Main chemical composition of untreated archaeological wood and reinforced archaeological wood 单位:%
2.4.2 傅里叶变换红外光谱分析
木栓脂、未处理考古木材和35%木栓脂加固考古木材的FT-IR光谱图如图5所示,各试样红外光谱图中官能团吸收峰的归属如表6所示。木栓脂区别于考古木材的主要特征峰为2 930和2 852 cm-1,归属于木栓脂脂肪族结构域[17],如图5中箭头指示,加固古木红外谱图中两个吸收峰的存在表明木栓脂加固剂进入考古木材细胞壁中。与未处理古木相比,加固古木在3 332和2 930 cm-1附近官能团的特征峰位向低波数方向偏移,可能是因为O—H和C—H的伸缩振动频率减小[23]。木栓脂加固古木中,与木质素相关的特征峰(如1 657,1 595,1 457 和1 135 cm-1)的峰强有所增加,表明加固古木中纤维素减少,木质素的相对含量增加,与表5中化学组分含量变化趋势一致。GC-MS结果表明,栓皮栎中提取的木栓脂单体主要为脂肪酸、二元羧酸和阿魏酸,其中脂肪酸类物质占木栓脂单体的54.96%,二元羧酸占9.62%,阿魏酸占4.58%。加固古木中O—H官能团伸缩振动减小,表明木材细胞壁上羟基与羧酸中的羧基发生了酯化反应[24]。

图5 木栓脂、未处理考古木材与35%质量分数木栓脂加固考古木材的红外光谱图Fig. 5 The FT-IR spectra of suberin, untreated archaeological wood, and 35% suberin reinforced archaeological wood

表6 未处理考古木材、加固考古木材与木栓脂红外波谱峰位归属及对应组分[17,25-29]Table 6 Peak position attribution, corresponding components and their FT-IR spectrum features of untreated archaeological wood, reinforced archaeological wood and suberin
结合电镜观察和化学组分分析,木栓脂加固剂作用于考古木材细胞壁的模式图如图6所示。

Ⅰ.细胞腔部分填充;Ⅱ.细胞腔完全填充;Ⅲ.沉积在细胞壁中注:图中黄色区域表示考古木材管胞横切面,蓝色表示木栓脂加固剂填充细胞腔,绿色表示木栓脂沉积在细胞壁中。
2.5 加固饱水考古木材吸湿性能
现代硬木松、未处理饱水古木和不同浓度加固液处理的古木在25 ℃时的平衡含水率变化如图7所示。在每一个相对湿度条件下,未处理饱水考古木材的平衡含水率(4.93%~15.73%)均高于现代材(4.42%~14.89%),这是由于饱水考古木材半纤维素、纤维素的降解,纤维素结晶度的减小以及无定形区比例相应增加的影响[30]。随着木栓脂加固剂引入浓度的增加,古木在每一个相对湿度下对应的平衡含水率均呈现下降趋势,当木栓脂质量分数为35%时,加固古木平衡含水率达到最低,平衡含水率变化范围为0.99%~5.76%,且明显低于未处理饱水古木的平衡含水率,吸湿性能变差。

图7 不同试样平衡含水率及其吸湿等温线GAB模型拟合曲线Fig. 7 The equilibrium moisture absorption of different specimens and their isotherm GAB model fitting curves
为了更深入地解析吸湿等温线,采用GAB模型对考古木材的吸湿等温线进行拟合,GAB模型可以反映试样在吸湿过程中平衡含水率和相对湿度的关系。图7中各试样吸湿等温线的GAB模型拟合曲线均为Ⅱ型“S”形。基于GAB模型得到的等温吸湿曲线参数见表7。从GAB模型拟合表中观察到,所有等温线模型拟合度良好(R2>0.97)。GAB模型中试验结果C的数值范围为2.676 59~12.178 49,均大于2;试验结果K的数值范围为0.778 32~0.978 15,均小于1。所有试样的C数值均远大于K的数值,表明单层的吸附热量高于多层的吸附热量[31]。在吸湿过程中,未处理饱水考古木材的单层分子饱和吸附量(0.055 32 cm3/g)比现代材(0.050 16 cm3/g)高10.29%,且木材有效比表面积与单层分子饱和吸附量成正比,表明饱水考古木材的降解使得木材细胞壁吸附作用有效比表面积变大。对比不同浓度木栓脂加固古木发现,总体上随着木栓脂加固剂浓度的增加,古木的单层分子饱和吸附量基本呈现下降趋势,木材细胞壁吸附作用有效比表面积也随之下降,从而吸湿性能降低,平衡含水率下降。
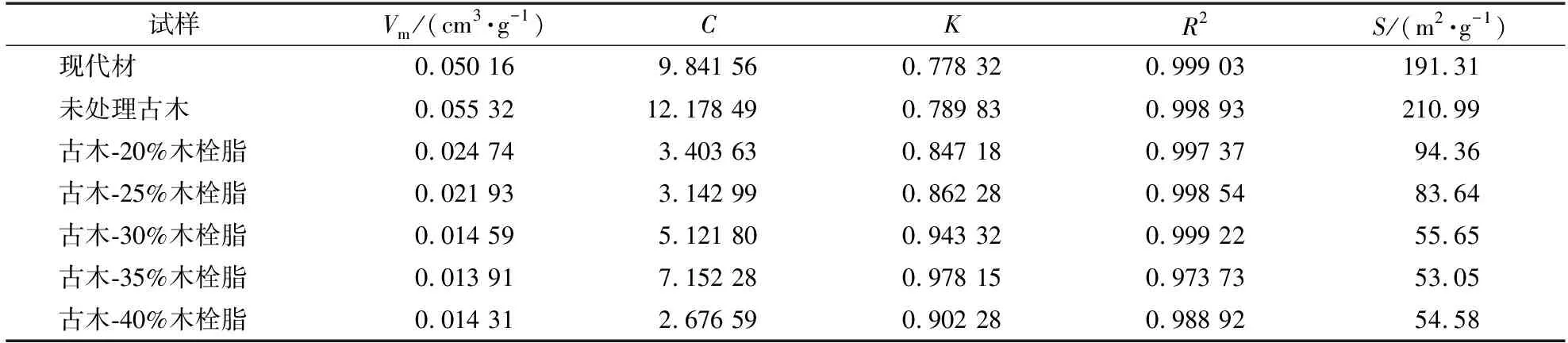
表7 不同试样吸湿等温线GAB模型参数Table 7 GAB model parameters of wood hygroscopic isotherms of different samples
吸湿性能的变化对木材尺寸稳定性有重要影响。在吸湿湿度范围内,0%~98%的相对湿度中,水分子主要存在于细胞壁内,水分子通过氢键与木材羟基发生作用[32]。与未处理古木相比,木栓脂加固古木单层分子饱和吸附量减少,表明木栓脂的憎水性阻碍了水分进入细胞壁,从而减弱了水分与木材细胞壁上羟基的作用,降低古木的吸湿性能。
3 结 论
1)经木栓脂加固处理的古木质量增加率、润胀率和抗收缩率增加,尺寸收缩率降低。当木栓脂加固剂质量分数为35%时,加固古木质量增加率、润胀率和各向尺寸抗收缩率达到最大值,各向尺寸收缩率达到最小值,古木尺寸稳定性提升效果最佳。结合微观形貌和化学成分分析结果表明,木栓脂加固剂不仅填充至考古木材管胞腔和纹孔中,而且沉积在木材细胞壁中,有助于提高古木的尺寸稳定性。
2)饱水考古木材在各相对湿度条件下的平衡含水率均高于现代材。经木栓脂加固处理后,古木在各相对湿度条件下的平衡含水率显著降低,表明加固古木的吸湿性降低。GAB模型拟合结果揭示了加固古木吸湿性降低的内在机理为木栓脂加固古木单层分子饱和吸附量减少,具有憎水性的木栓脂可阻碍水分进入细胞壁,从而减弱了水分与木材细胞壁上羟基的作用,降低古木的吸湿性能。
3)本研究中木栓脂与双癸基二甲基氯化铵表面活性剂配制的水溶性加固剂对提高考古木材的尺寸稳定性和降低古木的吸湿性能有较好的效果。随着木栓脂质量分数从0%增加到35%,加固考古木材质量增加率呈现上升的趋势,但更高浓度的加固剂对尺寸稳定性没有进一步提升效果。扫描电镜观察结果发现,加固剂在管胞腔内的填充不均匀,在今后的研究中将优化工艺条件,可以进一步改良木栓脂加固剂的分散度和均匀性,通过活化反应、乳化技术等调配渗透性更强的木栓脂复合加固剂,对饱水考古木材开展进一步高效且可逆的加固保护。
猜你喜欢
纺织标准与质量(2022年2期)2022-07-12 06:12:46
纺织标准与质量(2022年2期)2022-07-12 06:12:38
上海塑料(2021年3期)2022-01-06 14:05:02
山东冶金(2018年5期)2018-11-22 05:12:20
浙江工业大学学报(2017年5期)2018-01-22 02:03:42
广东农业科学(2017年5期)2017-08-29 10:37:54
宝藏(2017年5期)2017-07-18 11:54:21
材料科学与工程学报(2016年4期)2017-01-15 13:35:29
文物保护与考古科学(2016年4期)2016-05-17 05:31:17
食品工业科技(2014年9期)2014-03-11 18:15:40
