
综合分析和鉴定m6A RNA甲基化调节因子对前列腺癌进展及预后的影响
2023-12-01 08:56张乐宁静华张鑫王振刘虹汝张钰哲
中国医科大学学报 2023年11期
张乐,宁静华,张鑫,王振,刘虹汝,张钰哲,2,3
(1.大理大学基础医学院病理学与病理生理学教研室,云南 大理 671000;2.云南抗病原药用植物筛选重点实验室,云南 大理 671000;3.云南省昆虫生物医药重点实验室,云南 大理 671000)
前列腺癌是中老年男性常见的恶性肿瘤,已成为威胁人类健康的公共问题[1]。目前,前列腺癌的治疗方法包括手术、内分泌治疗、放化疗、冷冻治疗和免疫治疗等[2],但由于人们对前列腺癌的早期筛查不够重视,通常确诊时已为晚期或转移。前列腺癌患者即使进行积极的雄激素剥夺治疗,但由于长时间的药物治疗,患者会产生耐药性,从而演变为去势抵抗性前列腺癌[3]。因此,寻找更为有效的早期诊断标志物和治疗方案势在必行。
N6-甲基腺苷 (N6-methyladenosine,m6A) 是最常见的RNA甲基化修饰。m6A修饰与肿瘤的发生、分化、增殖、侵袭和转移有关[4-7]。本研究基于以往研究最广泛报道的13种m6A RNA甲基化调节因子 (以下简称m6A调节因子),利用癌症基因组图谱(The Cancer Genome Atlas,TCGA) 数据库中的RNA测序数据,系统分析了498例前列腺癌中这13种m6A调节因子的差异表达与临床病理特征的关联,评估m6A甲基化与前列腺癌进展和生存的相关性,为从甲基化修饰角度理解前列腺癌的发生和发展提供参考。
1 材料与方法
1.1 数据采集
从TCGA 数据库 (https://cancergenome.nih.gov/)中获取TCGA-PRAD数据,下载前列腺癌和癌旁样本的基因表达数据。排除同一患者的重复样本后,共有498例前列腺癌组织样本和52例正常前列腺组织样本。
1.2 m6A调节因子的选择
根据之前的研究[8-10],筛选出最广泛报道的13种m6A调节因子:METTL3、METTL14、WTAP、KIAA1429、RBM15、ZC3H13、YTHDC1、YTHDC2、YTHDF1、YTHDF2、HNRNPC、FTO、ALKBH5。提取13种基因的表达矩阵和临床样本的信息。
1.3 生物信息学分析
1.3.1 差异基因分析:为了研究m6A调节因子在前列腺癌中的作用,应用R语言 (4.2.1) limma包,分析13种m6A调节因子在前列腺癌和正常前列腺组织中的差异表达。
1.3.2 利用m6A调节因子在癌症中的表达水平进行无监督聚类分析:为了确定m6A调节因子是否与前列腺癌的预后相关,将498例前列腺癌样本进行分组。使用 R语言Consensus Cluster Plus包执行,根据集群和项目一致性原理,输出图形。集群数量通过累积分布函数曲线确定,为了验证根据13种m6A调节因子表达量进行的聚类分组在所有基因中的准确性,进行主成分分析,验证聚类分组效果。
1.3.3 m6A调节因子预后风险模型的构建:为了研究聚类分析与临床病理特征的相关性,从TCGA中下载前列腺癌患者的临床信息,使用limma包分析13种m6A调节因子在Cluster 1和Cluster 2中的表达情况,并研究这13种m6A调节因子与前列腺癌患者的临床病理特征之间的相关性。随后筛选出Cluster 1与 Cluster 2中的差异基因,对这些基因进行后续分析。利用基因本体论 (gene ontology,GO) 以及京都基因和基因组数据库 (Kyoto Encyclopedia of Genes and Genomes,KEGG) 富集分析,对差异基因进行功能注释。对差异基因进行单因素Cox回归分析,选择与生存密切相关的基因,应用LASSO Cox回归算法构建强大的预后特征,并计算每个基因的系数[11]。计算每例患者的风险评分,为每个基因评分的总和。根据前列腺癌患者风险评分的中位数,将前列腺癌分为高风险组和低风险组,绘制受试者操作特征曲线,计算曲线下面积 (area under the curve,AUC),获得预后特征的灵敏度和特异度。
1.4 风险因子的表达水平
进行单因素Cox回归分析,从297个差异表达基因中选出P< 0.01的基因,利用LASSO Cox算法选取误差最小的点,获得6个风险基因并构建风险模型。
1.5 基因表达和免疫浸润分析
使用癌症基因组分析平台 (http://bioinfo.life.hust.edu.cn/web/GSCALite/),对ROMO1在前列腺免疫微环境中的免疫浸润情况进行分析。为了探讨ROMO1的表达与前列腺癌免疫细胞的关系,使用癌症基因组分析平台中的Immune工具,分析ROMO1表达与前列腺癌免疫细胞浸润水平的关系。
2 结果
2.1 前列腺癌中m6A调节因子的关联性分析
与正常前列腺组织相比,前列腺癌患者中METTL3、METTL14、WTAP、KIAA1429、RBM15、ZC3H13、YTHDC1、YTHDC2、YTHDF1、YTHDF2、HNRNPC、FTO、ALKBH5的表达水平较高 (图1A、1B),超过半数 (11/13) 的m6A调节因子在前列腺癌中高表达。m6A调节因子与前列腺癌的TNM分期、年龄以及生存状态相关,尤其与T3、M0、N0和年龄≤65岁患者的临床特征显著相关 (图1C)。

图1 前列腺癌中m6A调节因子的分布Fig.1 Distribution of m6A regulators in prostate cancer
2.2 m6A调节因子的共识聚类鉴定2种不同的前列腺癌亚型
为了提供更为准确的分型,通过498例前列腺癌组织样本中13个m6A调节因子的mRNA 表达共识,将前列腺癌分为几个集群。聚类指数 k=2 时,是聚类间最大差异的最佳点。从m6A调节因子的表达相似性来看,选择一致累积分布函数 (cumulative distribution function,CDF) 坡度较小的曲线对应的k值为最佳。结果发现,k=3时,似乎具有较小的CDF值(图 2A、2B),说明将前列腺癌组织样本分为3个聚类最佳。但将其分为3组后,组间相关性较高,样本数量较少,因此分为2组 (Cluster 1和Cluster 2,图2C)。为了验证聚类的分组效果以及说明利用m6A调节因子进行的聚类分组在所有基因中的适用性,进行主成分分析。结果显示,可以将Cluster 1与 Cluster 2区分开来 (图2D),说明利用m6A调节因子的分类方法合理且准确。

图2 m6A调节因子一致聚类的识别Fig.2 Identification of consistent clusters of m6A regulators
2.3 通过共识聚类确定的类别与临床病理特征密切相关
在TCGA数据库下载前列腺癌患者的临床信息,对患者的分型和临床病理特征进行分析,发现大部分m6A调节因子在Cluster 1中高表达 (图3)。与Cluster 2相比,Cluster 1与诊断时的年龄 (≤65岁)、TNM分期 (T3、M0、N0)、生存状态 (存活) 显著相关,聚类结果与前列腺癌的恶性程度密切相关。
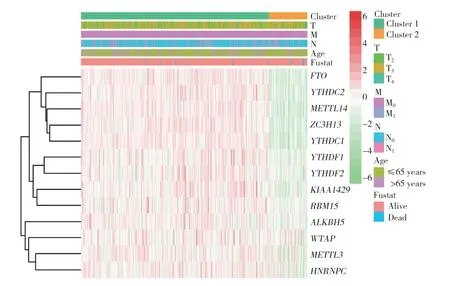
图3 Cluster 1、Cluster 2 与前列腺癌临床病理特征的关系Fig.3 Relationship between Clusters 1 and 2 and the clinicopathological features of prostate cancer
2.4 聚类间差异基因分析
从Cluster 1和Cluster 2中筛选出297个差异基因。GO分析结果表明,差异基因主要富集在恶性肿瘤的细胞基质黏附、黏附连接组织、线粒体呼吸链复合体组装、磷脂酰丝氨酸代谢过程、上皮细胞迁移的正向调节、细胞连接组织、氧化磷酸化作用等经典途径 (图4A、4B)。KEGG结果显示,差异基因富集于肿瘤的蛋白多糖、细胞外基质受体相互作用、转化生长因子-β信号通路、细胞骨架作用的调节、黏着斑、黏着连接 (图4C、4D)。

图4 利用生物过程GO分析和KEGG通路分析对Cluster 1亚群中表达较高的基因进行功能注释Fig.4 Functional annotation of highly expressed genes in the Cluster 1 subgroup using bioprocess GO and KEGG pathway analyses
2.5 m6A调节因子的风险特征与预后价值
从297个显著差异表达基因中,选出在单因素Cox回归分析中P< 0.01的基因,绘制森林图 (图5A)。风险比 (hazard ratio,HR) >1时,说明该基因是高风险基因;HR<1时,说明该基因是低风险基因。结果表明,SNORD60、SNHG25、SNORD104、C4orf48、OAS3、NME3、ROMO1、SNHG9、TMEM238、MRPL41、SNHG19这些基因均HR>1,说明以上基因表达水平较高时,前列腺癌患者的生存期较差。而SMIM22和MYOF均HR<1,说明这2种基因表达水平较高时,前列腺癌患者有更好的生存期。后续研究中,由这些基因代替m6A调节因子。

图5 6个m6A调节因子的风险特征Fig.5 Risk profiles of six m6A regulators
进行多因素Cox回归分析,利用LASSO Cox算法得到交叉验证的误差,选取误差最小的点并获得6个风险基因 (图5B),构建风险模型来预测样本的风险得分。为了研究风险模型预后作用,根据LASSO Cox算法得到的风险评分,利用风险评分的中位数,将TCGA数据集中的前列腺癌患者分为高风险组和低风险组,进行生存分析,结果显示,6个基因 (OAS3、MYOF、SMIM22、SNORD60、ROMO1、MRPL41) 构建的风险模型对前列腺癌的临床预后有较好参考价值 (P= 1E-07)。提示高风险组前列腺癌患者的生存期较差 (图5C)。
2.6 风险评分与前列腺癌的临床病理特征密切相关
6个风险基因与前列腺癌患者的年龄、分级、分期、T分期、M分期、N分期以及生存状态存在相关性 (图6A)。通过计算AUC,表明风险特征 (6个基因) 预测的准确率,AUC值越高,模型预测生存率越高。结果表明,风险评分可以预测前列腺癌的生存率 (AUC=0.727) (图6B)。单因素Cox回归分析结果显示,T分期、N分期、风险评分均可以作为高风险因素与风险评分相关 (图6C)。多因素Cox回归分析结果显示,风险评分可作为独立预后因子 (图6D)。以上证据表明,风险评分与前列腺癌临床病理特征密切相关,并且SNORD60、ROMO1、MRPL41与前列腺癌的恶性程度相关,特别是在高风险的前列腺癌中ROMO1表达水平更高。

图6 高低风险组中风险评分和临床病理特征的关系Fig.6 Relationship between risk scores and clinicopathological characteristics in high- and low-risk groups
2.7 ROMO1在前列腺癌中的免疫浸润情况
结果发现,ROMO1与前列腺癌的CD8+T细胞的免疫浸润相关性强 (图7),这也揭示了ROMO1与前列腺癌免疫浸润有一定关系。

图7 ROMO1在前列腺癌免疫微环境中的免疫浸润情况Fig.7 Immune infiltration by ROMO1 in the prostate cancer immune microenvironment
3 讨论
本研究结果显示,m6A调节因子的表达与前列腺癌的临床病理特征高度相关;使用6种基因构建的临床风险模型可以作为预后指标;ROMO1与高风险组前列腺癌的恶性程度相关性较高;ROMO1与前列腺癌CD8+T细胞的免疫浸润相关,其有望独立作为前列腺癌临床预后的指标。
ROMO1可以通过干扰活性氧的产生,影响细胞的状态[12]。通过释放细胞色素c激活细胞凋亡途径,最终导致细胞死亡,从而引发癌症。从正常细胞转化为癌细胞,ROMO1作为一种活性氧调节蛋白,已在多种肿瘤细胞中被发现,并在肿瘤的发生、发展中起作用[13]。有研究[14]发现,ROMO1在前列腺癌组织中高表达,且与前列腺癌的免疫细胞浸润水平和免疫途径相关,与本研究结果一致,说明了本研究结果的准确性。本研究发现,ROMO1与前列腺癌的CD8+T细胞间存在免疫浸润关系,这也许为前列腺癌的免疫疗法提供了新的治疗方案。在机体正常情况下,体内的T细胞可以通过识别异常的癌细胞,启动抗肿瘤的免疫反应,T细胞越多,人体发动免疫反应抵抗癌细胞的效果就越好。CD8+T细胞对肿瘤细胞具有较强的杀伤功能。因此,从免疫治疗的角度考虑,可能通过调节ROMO1的表达导致前列腺癌免疫细胞浸润的改变,从而影响前列腺癌微环境的改变,进而导致前列腺癌异质性,从而达到精准化个体治疗的目的。
综上所述,本研究结果系统展示了m6A调节因子在前列腺癌中的表达、潜在功能和预后价值。通过分析筛选出的目标基因ROMO1与前列腺癌恶性程度高度相关,有望作为前列腺癌临床预后的独立指标,为去势抵抗性前列腺癌的治疗提供理论基础。
猜你喜欢
青少年科技博览(中学版)(2022年11期)2023-01-07
汽车维修与保养(2021年8期)2021-02-16
家庭医学(下半月)(2020年3期)2020-05-30
家庭医学(下半月)(2020年3期)2020-05-30
家庭医学(下半月)(2020年3期)2020-05-30
中国生殖健康(2019年7期)2019-01-06
电子测试(2017年15期)2017-12-18
雷达学报(2017年6期)2017-03-26
工业设计(2016年4期)2016-05-04
电子设计工程(2015年6期)2015-02-27
