
桂枝甘草汤干预心肌缺血再灌注损伤大鼠心律失常的作用机制
2023-11-21 02:23朱明军唐进法曹英杰王小晓
中西医结合心脑血管病杂志 2023年21期
王 艳,高 原,朱明军,唐进法,李 彬,王 贺,曹英杰,崔 琳,王小晓,沈 思
心肌缺血再灌注(I/R)损伤是指在急性心肌梗死(acute myocardial infarction,AMI)发生后,药物或机械的早期再灌注在挽救缺血心肌的同时,会进一步导致心肌细胞功能障碍和组织损伤[1]。其中,心律失常是心肌I/R损伤的重要临床表现,以室性心律失常最为常见,严重者可发生恶性室性心动过速(ventricular tachycardia,VT)和心室纤颤(ventricular fibrillation,VF),是造成病人猝死的重要原因。细胞内钙超载和心肌能量代谢障碍是诱发心律失常的重要原因,而Na+-K+-ATP酶和Ca2+-Mg2+-ATP酶在维持细胞钙稳态及能量的产生和利用方面起重要作用[2-4]。心肌I/R发生时Na+-K+-ATP酶和Ca2+-Mg2+-ATP酶活性降低,导致心脏电活动的不稳定性和传导异常,增加心律失常发生率[5-6]。心肌细胞间信号传递的结构基础是缝隙连接(gap junction,GJ),在心脏电传导活动中发挥着重要的作用[7-8]。缝隙连接蛋白43(connexin 43,Cx43)则是心肌细胞间缝隙连接的主要连接蛋白,在心肌I/R状态下其表达含量和自身磷酸化状态的改变造成心肌细胞间电脱偶联,导致电冲动传导异常和心律失常的发生和持续[9-10]。桂枝甘草汤(Guizhi Gancao Decoction,GGD)温阳益气,是治疗心悸的基础方,出自汉代张仲景《伤寒论》,原文记载:“发汗过多,其人叉手自冒心,心下悸,欲得按者,桂枝甘草汤主之”。临床研究表明,GGD对不同类型心律失常具有较好疗效[11-12]。现有研究提示,GGD能够通过减轻钙超载、清除氧自由基、抑制炎症反应和细胞凋亡等机制对I/R心肌具有保护作用[13-14]。本研究通过构建大鼠心肌I/R损伤模型,从分子生物学层面研究GGD通过改善Cx43表达对心肌I/R致心律失常的影响及相关机制,为其减轻心律失常的临床治疗提供实验证据。
1 材料与方法
1.1 动物
Sprague-Dawley(SD)雄性大鼠90只,体质量250~300 g,购自河南省实验动物中心,动物许可证号为SCXK(豫)2017-0001。
1.2 药品及试剂
GGD浸膏(按照原著药量比例:即桂枝、甘草按2∶1的比例制备;1 g浸膏相当于9 g生药)由河南中医药大学第一附属医院提供。琥珀酸美托洛尔缓释片(Met)购自阿斯利康制药有限公司;血清肌酸激酶同工酶(CK-MB)、肌钙蛋白I(cTnI)检测试剂盒(批号:M3U4S6G5BR、78C8ABFAJY)购自Elabscience Biotechnology Co.,Ltd;Na+-K+-ATP酶、Ca2+-Mg2+-ATP酶试剂盒(批号:TF4X7MBJWY、24NWU56XV9)购自Elabscience Biotechnology Co.,Ltd;大鼠Cx43免疫组化试剂盒(批号:15386-1-AP)购自武汉三鹰生物技术有限公司;Kir2.1多克隆抗体(批号:B7301)、磷酸化Cx43(p-Cx43)多克隆抗体(批号:B7101)购自ImmunoWay Biotechnology Company;辣根过氧化物酶(HRP)标记山羊抗鼠二抗(批号:B0101)、HRP标记山羊抗兔二抗(批号:20000258)购自武汉三鹰生物技术有限公司;甘油醛-3-磷酸脱氢酶(GAPDH)抗体(批号:43929)购自GeneTex。
1.3 方法
1.3.1 造模方法
建立大鼠心肌I/R模型[15],末次给药1 h后,所有大鼠用10%水合氯醛(300 mg/kg,腹腔注射)麻醉后,仰卧位固定,连接PowerLab持续描记心电图Ⅱ导联。于颈部正中切开行气管插管并通过动物呼吸器辅助呼吸,在胸骨左缘第3肋与第4肋间间隙进行开胸手术,暴露心脏,用6-0号带针缝合线于左前降支(肺动脉圆锥与左心耳交界)处穿线,将一直径3 mm的乳胶管垫于结扎线与血管之间,系紧缝合线,进行心肌缺血30 min,然后取出垫扎乳胶管,进行再灌注120 min。对照组只穿线不结扎。缺血模型成功标准:结扎部位以下心肌组织变暗、发绀,心电图ST段明显抬高。再灌注模型成功标准:缺血区心肌发绀消失、逐渐变红,抬高的ST段逐渐回落>50%。
1.3.2 分组及给药方法
将90只大鼠随机分为对照组、I/R组、GGD低剂量组、GGD高剂量组和美托洛尔组,每组18只。GGD低剂量组、GGD高剂量组分别给予1.8、3.6 g/kg的GGD灌胃,每天1次,连续14 d;美托洛尔组给予美托洛尔9.5 mg/kg灌胃,每天1次,连续14 d;对照组及I/R组给予相同体积生理盐水。因麻醉或手术失败导致大鼠意外死亡,各组存活大鼠数量分别为对照组18只,I/R组、GGD低剂量组、美托洛尔组各15只,GGD高剂量组14只。
1.3.3 心电图指标及评分方法
观察再灌注后室性期前收缩(ventricualr premature contraction,VPC)、VT或VF的发生率和持续时间,并进行心律失常评分。根据1984年伦敦Lambeth会议确定的评分标准[16]:0分为无心律失常;1分为持续时间≤10 s的VT或其他心律失常,无VF;2分为持续11~30 s的VT或其他心律失常,无VF;3分为持续31~90 s的VT或其他心律失常,无VF;4分为持续91~180 s的VT或其他心律失常,和(或)少于10 s的可逆性VF;5分为持续超过180 s的VT或其他心律失常,和(或)超过10 s的可逆性VF;6分为不可逆性VF。
1.3.4 心肌组织Na+-K+-ATP酶、Ca2+-Mg2+-ATP酶水平的测定
采用分光光度法检测心肌组织Na+-K+-ATP酶、Ca2+-Mg2+-ATP酶的含量。制备心肌组织匀浆,用考马斯亮蓝和光谱法测定蛋白含量,比色定磷法测定,Na+-K+-ATP酶、Ca2+-Mg2+-ATP酶按照试剂盒的说明书进行测定。
1.3.5 血清CK-MB、cTnI水平测定
采用酶联免疫吸附试验(ELISA)法测定血清CK-MB、cTnI水平含量。再灌注120 min后腹主动脉取血,静置30 min后,以2 000 r/min离心15 min(取上清液)。按照ELISA试剂盒步骤测定血清中CK-MB、cTnI含量。
1.3.6 心肌组织的病理学观察
HE染色法观察心肌组织病理形态学改变。再灌注120 min后取出心脏,用冷的磷酸缓冲盐溶液(PBS)冲洗。取心肌组织转移至4%甲醛中浸泡,乙醇脱水,石蜡包埋切片(5 μm),用苏木精-伊红(HE)染色,在200倍放大镜下观察心肌组织的变化。
1.3.7 心肌组织Cx43表达的测定
采用免疫组化法检测心肌组织Cx43表达。取心肌组织转移至4%甲醛中浸泡,乙醇脱水,石蜡包埋切片(5 μm),脱蜡和抗原修复后,加3%的H2O2阻断内源性过氧化物酶,加一抗(1∶100),加酶标二抗,进行二氨基联苯胺(DAB)显色,苏木精复染,脱水和封片,镜检拍照显微镜下观察。
1.3.8 心肌组织中p-Cx43和Kir2.1的蛋白表达
采用蛋白免疫印迹(Western Blot)法测定心肌组织中p-Cx43和Kir2.1的蛋白表达。提取心肌组织总蛋白,聚丙烯酰胺凝胶电泳,转移至聚偏二氟乙烯膜,用脱脂奶粉封闭后,分别加p-Cx43抗体(1∶1 000)和Kir2.1抗体(1∶1 000),与GAPDH一抗(1∶1 000)平行进行孵育过夜,TBST冲洗,加HRP标记羊抗兔免疫球蛋白G(IgG)或HRP标记的山羊抗鼠IgG为二抗(1∶2 000),孵育1 h,应用化学发光试剂显色,使用Image J图像分析软件分析各蛋白条带的吸光度,蛋白表达水平以目的蛋白条带IOD值/GAPDH的IOD值表示。
1.4 统计学处理

2 结 果
2.1 GGD对心肌I/R大鼠心律失常的影响
用PowerLab连续监测心电图(见图1),分析再灌注期间心电图的变化,监测再灌注后心律失常发生率、持续时间和心律失常评分。结果显示,对照组大鼠在心电监测期间偶发VPC、手术时引起的VT,但无VF发生;其他组大鼠均发生VPC、VT或VF,且以VT及VF为主。与对照组相比,I/R组VPC次数明显增多,VT及VF持续时间明显延长,且心律失常评分明显增高(P<0.01),表明造模成功;与I/R组比较,GGD高剂量组、美托洛尔组VPC的发生次数明显减少,VT及VF的持续时间明显缩短,心律失常评分明显降低,差异均有统计学意义(P<0.05)。详见表1。
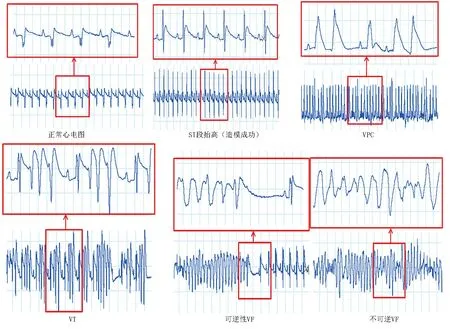
图1 PowerLab连续监测心电图示例
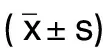
表1 各组VPC发生次数、心律失常持续时间、心律失常评分比较
2.2 GGD对心肌I/R损伤大鼠血清CK-MB、cTnI水平的影响
与对照组相比,I/R组大鼠CK-MB、cTnI水平明显升高(P<0.01);与I/R组相比,GGD低剂量组、GGD高剂量组和美托洛尔组CK-MB、cTnI水平明显降低(P<0.01)。详见图2、图3。
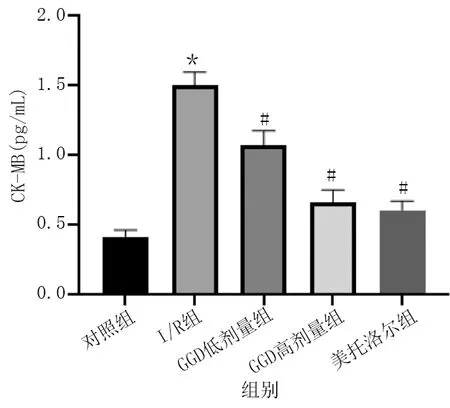
图2 各组血清CK-MB水平比较
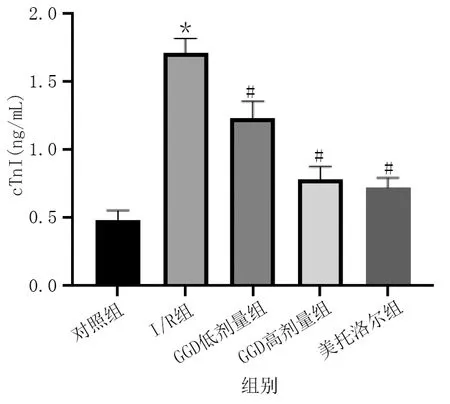
图3 各组血清cTnI水平比较
2.3 GGD对心肌I/R损伤大鼠心肌组织中Na+-K+-ATP酶和Ca2+-Mg2+-ATP酶水平影响
与对照组相比,I/R组大鼠Na+-K+-ATP酶和Ca2+-Mg2+-ATP酶活性明显降低(P<0.01);与I/R组相比,GGD低剂量组、GGD高剂量组和美托洛尔组Na+-K+-ATP酶和Ca2+-Mg2+-ATP酶活性明显升高(P<0.05或P<0.01)。详见图4、图5。

图4 各组心肌组织Na+-K+-ATP酶活性比较

图5 各组心肌组织Ca2+-Mg2+-ATP酶活性比较
2.4 GGD对心肌I/R损伤大鼠心肌组织作用的病理学改变
对照组心肌细胞结构清楚,心肌纤维细胞整齐,未见明显异常;I/R组心肌细胞排列紊乱,着色不均,心肌纤维肿胀、断裂;与I/R组比较,GGD低剂量组、GGD高剂量组和美托洛尔组心肌形态相对整齐,且随GGD剂量增加,心肌细胞内水肿和炎性细胞浸润明显减轻。详见图6。
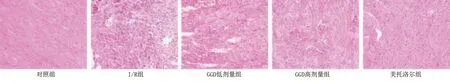
图6 各组大鼠心肌组织病理学变化(HE染色,×200)
2.5 GGD对心肌I/R损伤大鼠Cx43、p-Cx43和Kir2.1蛋白表达的影响
免疫组化结果显示:对照组可见较多棕黄色Cx43蛋白表达,分布规律,呈条带状;I/R组Cx43表达明显降低,呈点状无规律分布;与I/R组相比,GGD低剂量组、GGD高剂量组和美托洛尔组Cx43表达明显增多,分布较规律。详见图7。Western Blot结果显示:与对照组比较,I/R组p-Cx43和Kir2.1蛋白表达下降(P<0.01);与I/R组比较,GGD高剂量组和美托洛尔组p-Cx43和Kir2.1蛋白表达升高(P<0.05或P<0.01)。详见图8。

图7 免疫组化检测各组Cx43蛋白表达(×400)
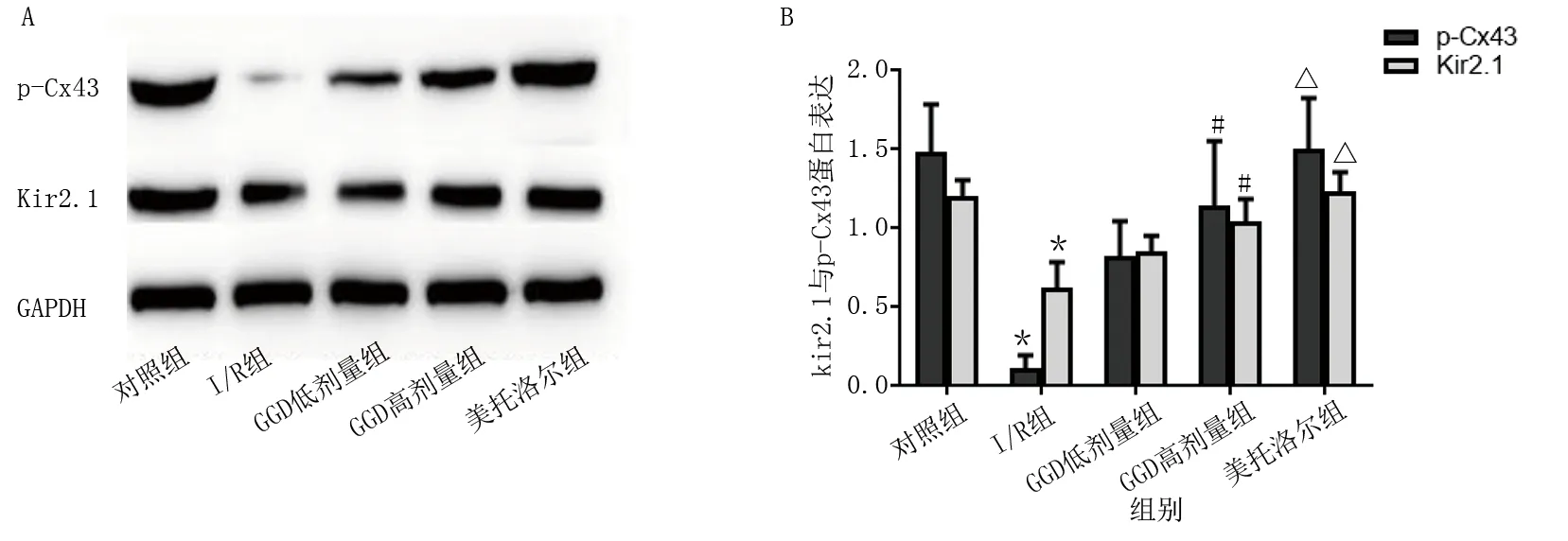
图8 Western Blot检测各组心肌组织中p-Cx43和Kir2.1蛋白水平
3 讨 论
GGD是张仲景治疗心悸的基础方,临床报道其具有很好的抗心律失常作用[11-12]。基础研究发现,GGD不仅能改善多种实验性心律失常[14,17],还可缩小心肌I/R大鼠心肌梗死面积从而发挥心肌保护作用,其机制涉及减轻钙超载、抗氧化、抑制炎症反应和细胞凋亡,调控TLR4/NF-κB信号转导通路等多个方面[13,18]。李冀等[19]发现桂枝甘草汤提取物30醇组分、水组分能够抗氯化钡、乌头碱及哇巴因等诱发的心律失常,机制可能与减少心肌细胞L型Ca2+、Na+通道电流及延长心肌细胞动作电位时程(APD)和抑制动作电位幅度(APA)有关。GGD乙酸乙酯部位的30%乙醇洗脱组分与肉桂酸协同对小鼠心律失常有一定的保护作用[20]。GGD及各组分含药血清均有抑制钙离子通道作用,该作用与钙离子阻滞剂相同[18]。本研究通过建立大鼠心肌I/R模型,探讨GGD对Cx43蛋白表达、Na+-K+-ATP酶活性、Ca2+-Mg2+-ATP酶活性、Kir2.1蛋白表达的影响。
心肌梗死病人发生心肌坏死、细胞膜通透性增加,使心肌细胞中CK-MB、cTnI等心肌损伤标志物大量释放入血,其升高水平与心肌细胞损伤程度关系密切[21-22]。本研究结果显示,I/R组大鼠血清CK-MB和cTnI水平较对照组明显升高,GGD药物组血清CK-MB和cTnI水平明显降低,HE染色结果也进一步表明GGD药物组较对照组可减轻心肌损伤程度,提示GGD药物组可改善大鼠心肌I/R损伤。
心肌I/R致心律失常的发生机制主要包括氧自由基堆积、钙超载、能量代谢障碍等几个方面。Na+-K+-ATP酶又称钠泵,其功能为水解ATP酶产生能量逆电化学梯度跨膜转运Na+和K+,维持细胞内外Na+、K+离子浓度梯度,Ca2+-Mg2+-ATP酶即为钙泵,可跨细胞膜主动将Ca2+转运到细胞外并主动摄取Ca2+从胞质中进入肌浆网。Na+-K+-ATP酶和Ca2+-Mg2+-ATP酶在维持细胞内钙稳态方面发挥着重要作用。当心肌I/R损伤发生时,心肌细胞能量代谢障碍,ATP生产减少,Na+-K+-ATP酶活力下降,细胞内Na+增多,K+减少,激活Na+/Ca2+交换使胞内Ca2+浓度增高,同时,Ca2+-Mg2+-ATP酶活性也下降,不能将胞内过多的Ca2+排出或摄入肌浆网,导致钙超载,诱发心律失常[23-25]。本研究结果表明,GGD药物组Na+-K+-ATP酶和Ca2+-Mg2+-ATP酶活性明显升高,有利于维持细胞内钙稳态,改善心肌细胞能量代谢,从而达到拮抗心肌缺血再灌注心律失常的作用。
缝隙连接通道分布于心肌细胞闰盘处,主要由3种连接蛋白构成,包括Cx40、Cx43、Cx45,其中心室肌的连接蛋白以Cx43为主,其正常表达及分布是保证心肌细胞间电耦联活动和协调心肌舒缩功能的关键因素[26]。生理状态下,Cx43有磷酸化和去磷酸化(Np-Cx43),但仅p-Cx43构成有功能的缝隙连接通道[27]。在心肌I/R损伤发生时,Cx43蛋白将表现脱磷酸化,间隙连接处的结构和功能变化,导致心肌组织阻力增加,导电性降低;间隙连接处的电性断开,导致电冲动的传导变慢,电活动合并,最终导致严重的室性心律失常。缝隙连接处的结构和功能变化,导致心肌组织阻力增加、电导性降低,出现缝隙连接的电脱耦连现象,导致电冲动传导变慢和折返性电活动的发生,最终导致严重的室性心律失常[28-29]。IK1的主要组成为Kir2.1,由KCNJ2基因编码,在维持细胞静息电位及钾离子电位平衡调节中发挥重要作用,是构成心肌细胞动作电位复极末期的主要电流通道之一[27,30]。在心肌缺血再灌注期间,心肌组织Kir2.1蛋白表达下调引起IK1下降,导致静息膜电位去极化,延长了动作电位时程,增加了心律失常风险[31-32]。本研究结果表明,I/R组Cx43蛋白的分布明显降低,p-Cx43和Kir2.1蛋白表达水平下降,GGD高剂量组、低剂量组Cx43表达明显增多,并在一定程度上逆转了p-Cx43和Kir2.1蛋白表达的下调,这可能是GGD改善心肌I/R心律失常的相关机制。
综上所述,GGD可减少VPC的发生次数,缩短VT及VF的持续时间,改善心律失常评分,其减轻心律失常的相关机制可能与改善Cx43的表达和磷酸化状态、提高Na+-K+-ATP酶和Ca2+-Mg2+-ATP酶活性及上调Kir2.1蛋白表达水平有关。通过提高ATP酶的活性,改善细胞能量代谢,维持心肌细胞内钙稳态,恢复细胞间缝隙连接蛋白功能与IK1电流,从而缩短动作电位时程与复极化进程,降低激动传导速度不均一,消除或减少折返电活动的发生,达到抑制心律失常的作用。
猜你喜欢
新乡医学院学报(2019年8期)2019-09-07
中国医药指南(2017年3期)2017-11-13
中国卫生标准管理(2015年2期)2016-01-14
中国继续医学教育(2015年1期)2016-01-06
天津医科大学学报(2015年2期)2015-12-22
肿瘤预防与治疗(2015年1期)2015-09-26
医学研究杂志(2015年11期)2015-06-10
中国当代医药(2015年21期)2015-03-01
中国卫生标准管理(2015年16期)2015-01-26
浙江大学学报(医学版)(2012年6期)2012-10-22
