
QTL analysis of early flowering of female flowers in zucchini(Cucurbita pepo L.)
2023-11-18 09:34:42QUShupingYANGDanYUHaiyangCHENFangyuanWANGKexinDlNGWenqiXUWenlongWANGYunli
Journal of Integrative Agriculture 2023年11期
QU Shu-ping, YANG Dan, YU Hai-yang, CHEN Fang-yuan, WANG Ke-xin, DlNG Wen-qi,XU Wen-long, WANG Yun-li#
1 Key Laboratory of Biology and Genetic Improvement of Horticultural Crops (Northeast Region), Ministry of Agriculture and Rural Affairs/College of Horticulture and Landscape Architecture, Northeast Agricultural University, Harbin 150030, P.R.China
2 Laboratory of Molecular Breeding of Pumpkin, College of Horticulture and Landscape Architecture, Northeast Agricultural University, Harbin 150030, P.R.China
Abstract Early flowering promotes early maturity, production, and the capacity to counteract biotic and abiotic stresses, making it an important agronomic trait in zucchini.The present study demonstrated that the zucchini inbred line ‘19’ consistently flowered early, taking significantly fewer days to bloom the first female flower (DFF) than the inbred line ‘113’.Genetic analysis revealed that DFF, an inheritable quantitative trait, is controlled by multiple genes.Based on the strategy of quantitative trait locus (QTL) sequencing (QTL-seq) combined with linkage analysis, three QTLs for DFF were identified on chromosomes 4, 11, and 20.This study used additional F2 populations grown under different environmental conditions for QTL mapping analysis of DFF with insertion/deletion (InDel) markers to validate these results.Using the composite interval mapping(CIM) method of R/qtl software, we only identified one major locus under all environmental conditions, located in a 117-kb candidate region on chromosome 20.Based on gene annotation, gene sequence alignment, and qRT-PCR analysis, we found that the Cp4.1LG20g08050 gene encoding a RING finger protein may be a candidate gene for the opposite regulation of early flowering in zucchini.In summary, these results lay a foundation for a better understanding of early flowering and improving early flowering-based breeding strategies in zucchini.
Keywords: Cucurbita pepo L., early flowering, days to bloom the first female flower, QTL analysis
1.lntro duction
CucurbitapepoL.is an economically important vegetable crop of the Cucurbitaceae family.Cultivars ofC.pepoare classified into eight horticultural groups: pumpkin, vegetable marrow, cocozelle, zucchini, scallop, acorn, crookneck,and straightneck.Among these groups, the zucchini group includes the highest valued vegetables worldwide(Formisanoet al.2012).Zucchini grows and reproduces simultaneously.It adapts to a wide range of environments,from tropical to temperate areas.Therefore, it is an ideal model for studying flowering time.
Flowering time is an important agronomic trait that contributes to early yield in annual plants, which determines the phase transition of a plant from the vegetative stage to the reproductive stage (Simpson and Dean 2002).Flowering time control is a complex life process essential for plant reproduction and growth.It is affected by environment,hormonal factors, and genetic factors (Sawaet al.2007).Early flowering allows plants to escape the biological and abiotic stresses exacerbated by long production cycles.Numerous genes involved in flowering time have been identified inArabidopsisthalianaand participate in the photoperiodic, vernalization, ambient temperature,aging, autonomous, and gibberellin pathways.It has been found that downstream genes that activate floral meristem identity are expressed in the shoot apex and leaves.These downstream genes includeFLOWERING LOCUST(FT) (Huanget al.2007),SUPPRESSOROF OVEREXPRESSIONOFCONSTANS1(SOC1) (Moonet al.2003; Liuet al.2008; Lee and Lee 2010),LEAFY(LFY),APETALA1(AP1),APETALA2(AP2),FRUITFULL(FUL),andCAULIFLOWER(CAL) (Henderson and Dean 2004).Moreover, these genes are involved in the signaling system in cucurbits (Linet al.2007; Luet al.2014; Zhouet al.2019).
In cucurbits, the early flowering of female flowers, which can effectively increase fruit production and advance the time to market, is an important goal of plant breeding.In cucumber, previous studies have identified a major quantitative trait locus (QTL) associated with early flowering(of either male or female flowers),Ef1.1, and a major QTL withlatefloweringlocus,Lf1.1.They also identified theFTgene as a possible candidate gene (Luet al.2014; Wanget al.2020).Based on map-based cloning and transgene technology, Wenet al.(2019) found thatTERMINAL FLOWER1(CsTFL1) controlled determinate growth and terminal flower formation, and the CsTFL1 protein competed with CsFT for interaction with CsNOT2a-CsFDP to inhibit these two traits.A major QTL,Qdff3-1, associated with days to female flowering was identified in watermelon, and theFTgene was identified as a major candidate gene (Gimodeet al.2019).Cucurbitamoschatais a cucurbit species responsive to inductive short-day (SD) photoperiods.Using zucchini yellow mosaic virus (ZYMV), it was found thatCmo-FTL1andCmo-FTL2expression inC.moschataserved as a florigenic signal that crossed the graft union in the phloem translocation stream (Linet al.2007).
Previous studies have identified several QTLs and genes associated with early flowering inC.pepo.Ethylene-related genes might be candidate genes responsible for early flowering, according to comparisons of single nucleotide polymorphisms (SNPs) unique to morphotypes performed using whole-genome resequencing (Xanthopoulouet al.2019).Two additional QTLs for early flowering were detected on LG9 and LG12 by genotyping-by-sequencing analysis of a recombinant inbred line (RIL) population (Montero-Pauet al.2017).The present study constructed an F2population by crossing the inbred lines ‘19’ and ‘113’.It also located three new QTLs associated with days to bloom the first female flower (DFF) based on the strategy of QTL sequencing(QTL-seq) combined with linkage analysis.Moreover, this study used additional F2populations grown under different environmental conditions for QTL mapping of DFF to validate the QTL-seq results.It also identified the candidate genes associated with the early flowering of female flowers.The results herein reveal a new locus associated with early flowering that can be used to design QTL pyramiding strategies in the zucchini molecular breeding system.
2.Materials and methods
2.1.Plant materials
TheC.pepoinbred line ‘19’ showed significantly earlier flowering of the first female flower than the inbred line‘113’.F1, F1´, and F2populations derived by crossing ‘19’and ‘113’ were constructed to analyze the inheritance and map QTLs for DFF.In this study, 40 ‘19’, 40 ‘113’, 40 F1and 580 F2plants were planted in greenhouses in April 2019 (2019G); 40 ‘19’, 40 ‘113’, 40 F1, 40 F1´ and 480 F2plants were planted under open cultivation in May 2020(2020O); and 40 ‘19’, 40 ‘113’, 40 F1, 40 F1´ and 195 F2plants were planted in greenhouses in April 2020 (2020G).All plants were grown at the Xiangyang Base of Northeast Agricultural University, Harbin, China (45°77´N, 126°92´E).All individuals were managed using standard horticultural procedures (irrigation, hand weeding, and hand pollination).The days from emergence to flowering of the first female flower on each plant were counted as DFF.Under each environmental condition, the DFF values of ‘19’, ‘113’ and F1(20 plants per family) were determined, and the mean DFF values were calculated.DFF data were collected from F2individuals in 2019G, 2020O, and 2020G for QTL analysis.
2.2.DNA pool construction and QTL-seq
Genomic DNA was extracted from individual ‘19’, ‘113’, F1,and F2plants using the cetyltrimethylammonium bromide(CTAB) method (Murray and Thompson 1980) for QTL-seq.The early DFF pool (E-pool) and late DFF pool (L-pool)were constructed by mixing equal amounts of DNA from 30 extremely early flowering plants (at 27 days to anthesis(DTA) or less) and 30 extremely late flowering individuals(at 38 DTA or more) from the 2019G F2population.The parental DNA pools were constructed by mixing equal amounts of DNA from 30 ‘19’ and 30 ‘113’ plants.A paired-end sequencing library with approximately 350-bp DNA reads was constructed from the four pools using the Illumina HiSeq 2000 platform through a commercial service at BioMarker (Beijing, China).Raw reads were mapped to the reference genome of zucchini (http://cucurbitgenomics.org/organism/14, Montero-Pauet al.2018).The output of paired-end alignment files was compared with that of BWA Software (Li and Durbin 2009) to filter clean reads.SNPs and insertions/deletions (InDels) among ‘19’, ‘113’, the E-pool, and the L-pool were detected using GATK Variant Analysis Software (McKennaet al.2010).The SNP/InDel index and the Δ(SNP/InDel-index) were calculated for all positions to identify candidate regions associated with DFF (Abeet al.2012).SNP-index and InDel-index graphs were generated for the E-pool and L-pool by plotting the average SNP-index and InDel-index values, respectively.According to the null hypothesis, 99% confidence intervals were selected to identify the candidate regions harboring QTLs for the DFF trait.
2.3.QTL mapping with lnDel markers
Polymorphic InDel markers were developed to validate the results of QTL-seq analysis in the candidate regions of DFF based on the InDels between ‘19’ and ‘113’.InDel markers were designed with Primer Premier 5.0 and are listed in Appendix A.PCR was carried out using 10 μL samples containing ~40 ng of genomic DNA, each primer at 0.5 μmol L–1, 200 μmol L–1dNTPs, 1× reaction buffer, and 0.5 U ofTaqDNA polymerase (Aidlab Biotechnologies, Beijing,China).PCR amplification was performed with the following program: 94°C for 5 min; 35 cycles of 94°C for 30 s, 56°C for 30 s, and 72°C for 30 s; and 72°C for 5 min.Products were separated on an 8% polyacrylamide gel by electrophoresis.After electrophoresis at 220 V for 2 h, the gel was stained with 0.3% AgNO3solution, and the silver-stained DNA bands were revealed.
2.4.Data analysis
Genetic mapping of the F2individuals was carried out with InDel markers on the target chromosome using JoinMap 4.0.The recombination values were converted into map distances expressed in centiMorgan (cM) using the Kosambi mapping function.A, B, and H represent homozygous‘19’, homozygous ‘113’, and heterozygous genotypes,respectively.QTL analysis was performed in the R/qtl Software package (http://www.rqtl.org/) with the composite interval mapping (CIM) method following Wenget al.(2015).The significance of each QTL interval was tested by a likelihood-ratio statistic (LOD).The LOD threshold for declaring QTLs was established separately for each environment using 1 000 permutations at a significance threshold of 0.05.For each detected QTL, a 2-LOD support interval was calculated and defined by left and right markers.The QTLs were named according to the different environments and chromosomes.
2.5.Sequencing and functionals annotation of predicted genes
Gene ID, function, and structure data were obtained from the Cucurbit Genomics Database (Montero-Pauet al.2018).SoftBerry Online Software (http://linux1.softberry.com/) was used for dicot plant (Arabidopsis)-based gene structure prediction.The functions and motifs of candidate genes were retrieved from the NCBI website (http://archive-dtd.ncbi.nlm.nih.gov/).The critical elements of the promoter sequence were analyzed by the PlantCARE website (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/).
2.6.RNA isolation and qRT-PCR
To determine the expression of candidate genes, shoot apices of ‘19’ and ‘113’ at the 4-leaf, 7-leaf, and 10-leaf stages were collected.Total RNA was extracted using TRIzol reagent (Invitrogen, USA).Dried RNA samples were dissolved in ddH2O and treated with diethylpyrocarbonate.First-strand cDNA was prepared according to the PrimeScript RT Reagent Kit with the gDNA Eraser (TaKaRa, Kyoto,Japan) protocol.Four primer pairs for candidate genes and three homologous genes involved in early flowering were designed (Appendix B).Taq SYBR Green qPCR Premix(Yugong Biolabs, Inc., CN) was applied to perform qRT-PCR.The program used in this assay was as follows: 96°C for 1 min; 30 cycles of 95°C for 15 s, 58°C for 15 s, and 72°C for 45 s.TheActingene was used as the internal reference.Three technical replicates were set for each sample, and relative expression levels were quantified using the 2–ΔΔCTmethod (Livak and Schmittgen 2001).
3.Results
3.1.Phenotypic and inheritance analyses of the DFF trait
This study determined the DFF and the node of the first female flower for 20 randomly selected ‘19’ and ‘113’ plants grown under three different environmental conditions.In 2019G, the DFF of ‘19’ was 28 DTA, and the DFF of ‘113’ was 39 DTA.In 2020O, the mean DFF of ‘19’ was 36 DTA, and the mean DFF of ‘113’ was 41 DTA.In 2020G, the mean DFF of ‘19’was 36 DTA, and the mean DFF of ‘113’ was 42 DTA (Table 1).The DFF of ‘19’ included approximately 5–11 DTA more than that of ‘113’, illustrating that ‘19’ was an early-flowering line,‘113’ was a late-flowering line, and ‘19’ was significantly different from ‘113’ in terms of flowering under all the three environmental conditions.The first female flower on the main vine in ‘19’ occurred at node 9, and the first female flower on the main vine in ‘113’ occurred at node 12 (Fig.1).F1plants flowered on the same day as the parent ‘19’, illustrating that early flowering was a dominant trait.The DFF of the F2population showed continuous variation and bidirectional transgressive segregation in the three environments,suggesting that DFF was a quantitative trait under polygenic control and that there was a genetic difference in flowering time between ‘19’ and ‘113’ (Table 1; Fig.2).
3.2.QTL mapping of the DFF trait with QTL-seq
To map the candidate regions associated with DFF, we used the QTL-seq approach to identify the candidate QTLs.Four samples (‘19’, ‘113’, E-pool, and L-pool) of DNA were sequenced to obtain a total of 57.16 Gb clean bases.A total of 71.4 and 54.6 million clean reads were obtained from the E-pool and L-pool, respectively, by QTL-seq.The clean reads from the E-pool and L-pool had high (43× and 33×) average read depths, and the Q30 values reached 91.59 and 89.75%, respectively.The percentages of clean reads from the E-pool and L-pool mapped to the reference genome of zucchini were 98.23 and 98.17%, which resulted in 10× coverage ratio of 94.16 and 92.66%, respectively.These results indicated that the sequencing results were reliable for gene mapping.A total of 54 382 high-quality SNPs and 29 095 high-quality InDels were obtained on all 20 chromosomes.Δ(SNP-index) and Δ(InDel-index) were calculated and plotted by comparing the SNP-index and InDel-index results of the E-pool and L-pool at the genomic positions.The threshold of the Δ(SNP-index) value was 0.25; it was identified that the Δ(SNP-index) values of three regions (with a total length of 2.11 Mb on chromosomes 4,11, and 20) were greater than the threshold value (Fig.3-A).The threshold of the Δ(InDel-index) value was 0.24; the Δ(InDel-index) values of four regions (with a total length of 2.11 Mb on chromosomes 4, 4, 11, and 20) were greater than the threshold value (Fig.3-B).Regarding the intersection of two association regions, four candidate regions with a total length of 2.04 Mb on chromosomes 4, 4, 11, and 20 were associated with DFF, containing a total of 280 genes(Appendix C).There was 0.02 Mb between the two regions on chromosome 4.Therefore these two regions could be the same locus, which we named the QTLsCpDFF4.1,CpDFF11.1andCpDFF20.1.
3.3.lnDel markers narrowed down a major QTL interval
To confirm the DFF QTLs detected by QTL-seq, we conducted a QTL analysis of 554 F2plants from 2019G,462 plants from 2020O, and 191 plants from 2020G.On chromosomes 4, 11, and 20, a total of 42 polymorphic InDel markers were selected between ‘19’ and ‘113’.QTLs that could explain more than 10% of the phenotypic variations were considered major-effect QTLs.Information on the QTLs is provided in Table 2 and Fig.4.In 2019G,the LOD threshold was 3.32, and one major-effect QTL,Cp19G20.1, was detected and located on chromosome 20.The QTLCp19G20.1was delimited by two markers,Chr20_7444755 and Chr20_8065035, and the peak marker was Chr20_7855309 (Fig.4-A).The LOD score ofCp19G20.1was 13.88, and this QTL could explain 10.57% of the phenotypic variation.The maximin LOD score on chromosome 4 was 2.22 and peaked at marker locus Chr04_33799, located in theCpDFF4.1region using the QTL-seq method (0–0.58 Mb on chromosome 4).The maximin LOD score on chromosome 11 was 3.21 and peaked at marker locus Chr11_160639 in 2019G, located in theCpDFF11.1region using the QTL-seq method(0–0.59 Mb on chromosome 11).
In 2020O, the LOD threshold was 3.30, and one majoreffect QTL,Cp20O20.1, was detected on chromosome 20, which delimited it to the same region asCp19G20.1(Fig.4-B).The LOD score ofCp20O20.1was 11.30, and this QTL could explain 10.80% of the phenotypic variation.The maximin LOD score on chromosome 4 was 1.89 and peaked at marker locus Chr04_33799, located in theCpDFF4.1region.The maximin LOD score on chromosome 11 was 1.67 and peaked at marker locus Chr11_3049065, located outside theCpDFF11.1region.
In 2020G, the LOD threshold was 3.49, and one major-effect QTL,Cp20G20.1, was detected on chromosome 20,which was delimited to the same region asCp19G20.1andCp20O20.1(Fig.4-C).The LOD score ofCp20G20.1was 5.26, and this QTL could explain 12.59% of the phenotypic variation.The maximin LOD score on chromosome 4 was 2.28 and peaked at marker locus Chr04_11010856, located outside theCpDFF4.1region.The maximin LOD score on chromosome 11 was 1.60 and peaked at marker locus Chr11_8520572, located outside theCpDFF11.1region.Cp19G20.1,Cp20O20.1, andCp20G20.1showed negative additive effects, indicating that the alleles increasing DFF were contributed by ‘113’.
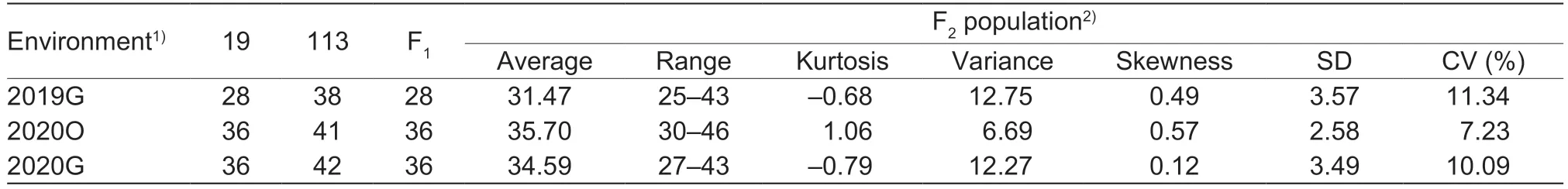
Table 1 Descriptive statistics for days to bloom the first female flower (DFF) trait among the parents and F2 population
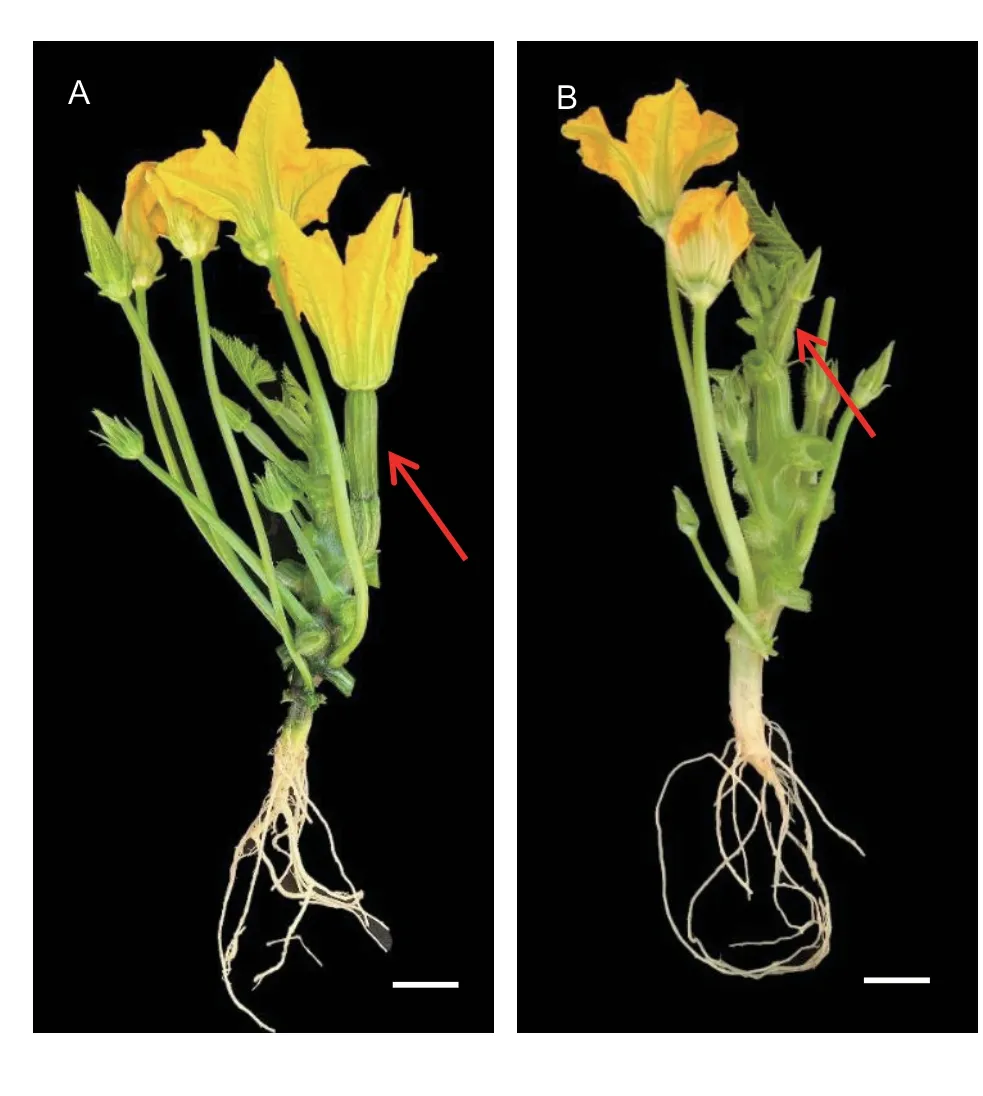
Fig.1 Flowering time phenotype in the two adult parents ‘19’(A) and ‘113’ (B).The red arrows indicate female flowers in‘19’ and ‘113’.Scale=5 cm.
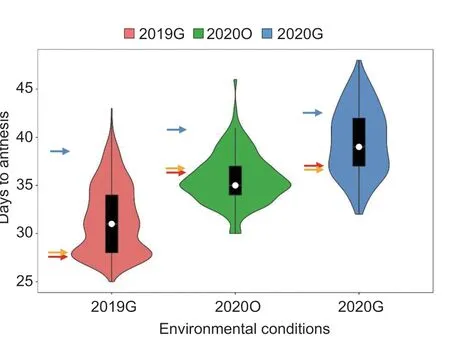
Fig.2 The frequency distribution of the F2 population.The red arrows indicate flowering time phenotype in ‘19’, the blue arrows indicate flowering time phenotype in ‘113’, and the orange arrows indicate flowering time phenotype in F1.2019G,2020O, and 2020G, planted in greenhouses in April 2019, under open cultivation in May 2020, and in greenhouses in April 2020,respectively.
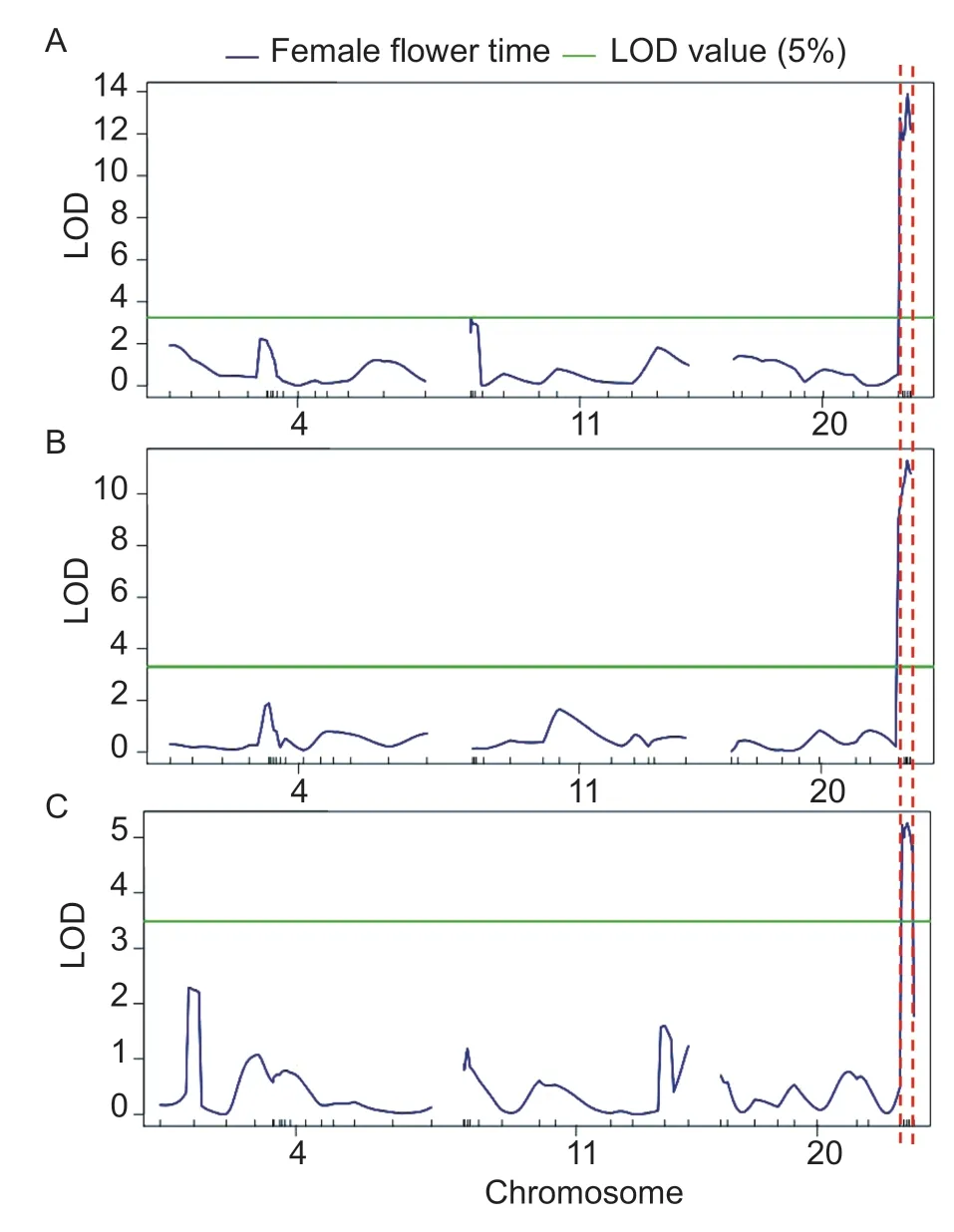
Fig.4 Genetic mapping of days to bloom the first female flower(DFF) trait by Indel markers.A, genetic mapping of DFF trait in 2019G.Using 554 individuals of F2 from ‘19’ב113’, one QTL was identified on chromosome 20.B, genetic mapping of DFF trait in 2020O.Using 462 individuals of F2 from ‘19’ב113’, one QTL was identified on chromosome 20.C, genetic mapping of DFF trait in 2020G.Using 191 individuals of F2 from ‘19’ב113’,one QTL was identified on chromosome 20.2019G, 2020O,and 2020G, planted in greenhouses in April 2019, under open cultivation in May 2020, and in greenhouses in April 2020,respectively.LOD, likelihood-ratio statistic.
Regarding the intersection of associated regions from QTL mapping using the CIM method in three environments and QTL-seq analysis, a major QTL,CpDFF20.1, was mapped in a 620.28-kb interval on chromosome 20 between the markers Chr20_7444755 and Chr20_8065035.Among the 2019G F2individuals, extremely early flowering(DTA<28) and extremely late flowering (DTA>39) plants were selected to analyze InDel haplotypes on the end of chromosome 20, and theCpDFF20.1locus was narrowed down to an interval between the markers Chr20-7737657 and Chr20-7855309 (Appendix D).Therefore, the 117.65 kb genomic region harbored the candidate genes for DFF.
3.4.Candidate genes analysis of the important QTL
According to the Cucurbit Genomics Database and SoftBerry,a total of 17 putative genes in the 117.65 kb QTL region were identified.The positions, InDels, SNPs, and predicted functions of these genes are shown in Appendice D and E.SNPs and InDels located in promoter elements, untranslated regions (UTRs), and coding sequence (CDS) regions were analyzed between the two parental lines.Among the 17 genes, four (Cp4.1LG20g08030,Cp4.1LG20g08040,Cp4.1LG20g08050, andCp4.1LG20g08110) had nonsynonymous changes in the CDS region or in the promoter element between ‘19’ and ‘113’, and the functions of these four genes were related to flowering date.
This study collected young leaves from ‘19’ and ‘113’ to determine the expression levels of the candidate genes,before female flower differentiation (the 4-leaf stage), at female flower differentiation of the early-flowering line (the 7-leaf stage), and at female flower differentiation of the lateflowering line (the 10-leaf stage) (Fig.5).The results showed that the expression level ofCp4.1LG20g08110at the 7-leaf stage in the early-flowering line ‘19’ was significantly lower than that in the late-flowering line ‘113’; in contrast, the expression level ofCp4.1LG20g08110at the 10-leaf stage showed the opposite pattern between the early- and late-flowering lines.Upon comparing the expression levels ofCp4.1LG20g08030andCp4.1LG20g08040, which were different between the early-flowering line ‘19’ and the late-flowering line ‘113’, only one stage showed significantly different expression levels.The expression level ofCp4.1LG20g08050in the earlyflowering line ‘19’ was significantly lower than that in the lateflowering line ‘113’ at the three key stages (the 4-leaf, 7-leaf,and 10-leaf stages).Cp4.1LG20g08050encodes C3HC4-type RING finger protein that may be a regulator of the cold signaling pathway.This result indicated that the expression level ofCp4.1LG20g08050was negatively correlated with the early flowering time at key periods in flowering induction.Cp4.1LG20g08050contain RING conserved domain of c-Cbl,which is intrinsic to E3 ubiquitin-protein ligase activity.E3 ligase is involved in seed dormancy, flowering timing, defense response, and salt stress regulation in plant development.There is one nonsynonymous SNP between the two parental lines in the first exon ofCp4.1LG20g08050(Appendix F).Changing amino acids between ‘19’ and ’113’ might lead to gene expression and be involved in flowering timing.
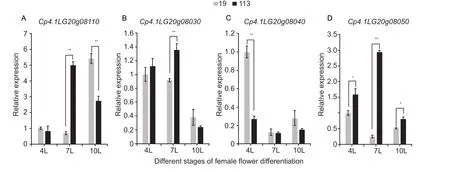
Fig.5 Relative expression levels of four candidate genes in ‘19’ and ‘113’ at different stages of female flower differentiation.A,expression levels of Cp4.1LG20g08110.B, expression levels of Cp4.1LG20g08030.C, expression levels of Cp4.1LG20g08040.D, expression levels of Cp4.1LG20g08050.For the two parents, the expression level at the 4-leaf stage (4L) in ‘19’ was set to a value of 1 and used as a reference, respectively.Each was repeated three times, and 15 plants were mixed in equal amounts to form one replicate in two pools.Data are mean±SD (n=3).** indicates an extremely significant difference, P<0.01; * indicates a significant difference, P<0.05.
To elucidate the molecular mechanism underlying flowering time variation in zucchini, we investigated the expression of the flowering-related genes,CpFT,CpSOC1,andCpAP1(Fig.6).The expression levels ofCpSOC1andCpAP1in the early-flowering line ‘19’ were significantly higher than those in the late-flowering line ‘113’ in all three stages.Significant differences inCpFTexpression were detected only before the first female flower bloomed.These results indicated that the expression levels ofCpSOC1andCpAP1were positively associated with early flowering time.
4.Discussion
Arabidopsisand rice show a clear separation of the vegetative and reproductive phases.Unlike classic model plants,zucchini exhibits leaves produced from the flank of the shoot apical meristem, and male or female flowers are produced from the axils of the main stem.Zucchini also shows no clear distinction between the vegetative and reproductive phases.InC.pepo, early flowering could be desirable until full fruit maturation in short growing seasons, and sink competition is achieved between developing leaves and developing fruits until a balance in nutrients.Growing reproductive organs,such as developing fruits and seeds, may act as dominant sinks, thereby limiting further vegetative growth (Ogdenet al.2020).In this study, we noticed that female flowers of ‘19’ and‘113’ bloomed before male flowers in the long day condition(spring), and male flowers bloomed before the female flowers in the short day condition (fall).It is demonstrated that low temperature and long-day environment might induce earlier differentiation of female flowers and early flowering of female flowers.Compared with ‘113’, ‘19’ line exhibited highly consistent and stable early female flowering in both long and short day conditions.The node of the first female flower was also counted in ‘19’ and ‘113’ with 20 per plant.The first female flower appeared at the 9th node in ‘19’ and the 13th node in ‘113’, which indicated that the first female flowers of‘19’ differentiated earlier than that of ‘113’.We counted days to bloom the first male flower, ‘19’ line also exhibited stable early male flowering in both long and short day conditions, which indicated that genes associated with DFF also influenced the differentiation and development of male flowers.
We used the highly diversified parents ‘19’ and ‘113’ to establish a mapping population to examine the flowering time of the first female flower.The F2population showed wide segregation for the phenotypic traits, which indicated that the population was suitable for mapping.The hybrid F1generation showed stable early female flowering, which was consistent with the phenotype of ‘19’.Thus, ‘19’ is a good germplasm resource with the potential to serve as an early-flowering gene donor in zucchini crossbreeding.We also observed that the DFF of ‘19’ and ‘113’ in 2019G (28 DTA and 39 DTA) was noticeably less than that in 2020O and 2020G environments (36 DTA and 41 DTA in 2020O,36 DTA and 42 DTA in 2020G).In the F2population, the average in 2019G was also significantly less than that in 2020G and 2020O.After plant sprouts, the average daily minimum temperature in May and June of 2019 and 2020 is 9 and 15°C, respectively.It is demonstrated that environmental conditions such as low temperature could effectively influence DFF in zucchini.
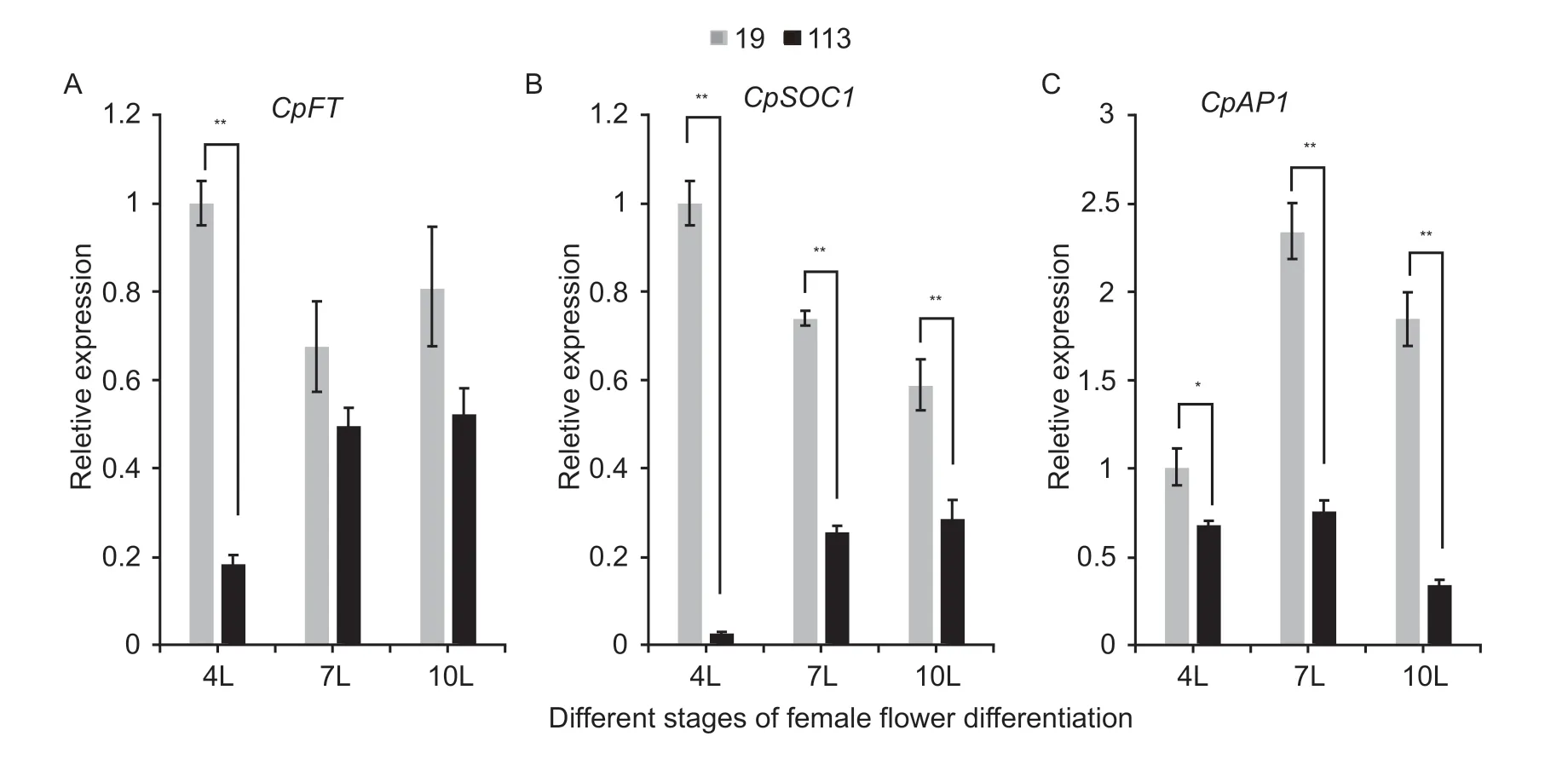
Fig.6 Expression levels of key genes in ‘19’ and ‘113’ at different stages of female flower differentiation.A, expression levels of CpFT.B, expression levels of CpSOC1.C, expression levels of CpAP1.For the two parents, the expression level at the 4-leaf stage (4L) in ‘19’ was set to a value of 1 and used as a reference, respectively.Each was repeated three times, and 15 plants were mixed in equal amounts to form one replicate in two pools.Data are mean±SD (n=3).** indicates an extremely significant difference, P<0.01; * indicates a significant difference, P<0.05.
The molecular markers and candidate genes related to flowering time in zucchini are less well known than those in other cucurbit species, such as melon and cucumber.A saturated genetic map was constructed using genotypingby-sequencing analysis of a RIL population.Based on this, one major QTL for early flowering was detected at the head of chromosome 12, and one minor QTL was detected at the end of chromosome 9 (Montero-Pauet al.2017).Whole-genome resequencing ofC.pepomorphotypes was performed.Among SNPs uniquely associated with DFF, several genes involved in early flowering among the various morphotypes ofC.peposubspecies were selected(Xanthopoulouet al.2019).These genes were located on the end of chromosome 4, in the middle of chromosome 17, at the end of chromosome 19, and at the head of chromosome 20.Homologous regions of major QTLs controlling early flowering in cucumber were located on different chromosomes or at different locations (the head of chromosome 3, the middle of chromosome 10, the middle of chromosome 18, and the middle of chromosome 19 in the zucchini genome), in contrast to published QTLs in cucumber (Luet al.2014; Wenet al.2019; Wanget al.2020).The Gene Expression Atlas for theC.pepo(CupeGEA)was developed, and based on gene expression analysis,several flowering-related genes related to early flowering were selected.These genes encoded APETALA-2, EARLY FLOWERING 3 and 4, Flowering Control protein (FCA), FT protein, and flowering time control FPA-like protein detected on chromosomes 5, 10, 14, 16, 18, and 19 (Xanthopoulouet al.2021).Our study used QTL-seq combined with linkage analysis to sequence four pools (‘19’, ‘113’, E-pool, and L-pool) of DNA from the 2019G F2population, mapping the candidate regions associated with DFF.Three QTLs were identified at the head of chromosomes 4 and 11 and the end of chromosome 20 (Appendix C), which were different from the published regions in cucumber and zucchini.Furthermore, ethylene-related genes involved in signaling and synthesis were not found in the major QTL region on chromosome 20.Identifying and cloning more genes controlling flowering time could better explain the molecular mechanism of early flowering and provide important theoretical and technical support for zucchini breeding.
This study performed QTL analysis to confirm the DFF QTLs detected by QTL-seq, using data from three environment conditions.The results indicated that the environment seldomly affected the major-effect QTLs, and minor-effectCpDFF4.1andCpDFF11.1were unstable and easily affected by the environment.Conventional breeding techniques require the selection of phenotypes for stable yield and the evaluation of yield in multiple environments,which can be time and labor-intensive.Marker-assisted selection (MAS) is a powerful tool for selecting target traits with high precision that are not affected by the environment, which effectively accelerates the breeding process.High-resolution mapping of QTLs may be used to develop reliable markers for MAS (<5 cM away from the genes/QTLs).Utilizing MAS based on DFF would potentially aid in selecting early flowering, which would shorten production time and reduce costs.It is essential to develop polymorphic molecular markers associated with DFF for the application of molecular breeding in zucchini.Our study detected the major QTLCpDFF20.1in all three environments, which could explain 10.57–12.59% of the phenotypic variation.Finally,CpDFF20.1was flanked by the markers Chr20_7737657 and Chr20_7855309 in a 2.5–4.9 cM region.These two markers would be extremely useful in MAS for rapidly preparing early-flowering and high-yielding hybrids with different genetic backgrounds.
In the major QTL region, four genes had nonsynonymous CDS or promoter element changes between ‘19’ and ‘113’.According to the qRT-PCR results, the expression level of onlyCp4.1LG20g08050was strongly negatively correlated with early flowering at three key periods in flowering induction.Cp4.1LG20g08050contains the RING finger motif, which can be classified as a C3HC4-type RING,similar to theHOS1gene inArabidopsis(Leeet al.2001).It was reported that thehos1mutant exhibited a low level ofFloweringLocusCexpression and early flowering.TheHOS1gene is also a key negative regulator of low temperature-responsive gene transcription and regulates downstream cold-responsive gene expression.Based on the same structural characterization and expression patterns of these two genes, it is deduced thatCp4.1LG20g08050might be the key gene negatively correlated with early flowering in zucchini, and it might be influenced by temperature in the planting environment.
For floral induction, certain numbers of young leaves are required to perceive relevant environmental signals and induce flowering-related gene expression (Shivashankara and Mathai 1995).Young leaves of the early-flowering zucchini line ‘19’and the late-flowering line ‘113’ were selected for expression ofCpFT,CpSOC1, andCpAP1at key periods of flowering induction.There were no significant differences between ‘19’and ‘113’ inCpFTexpression at female flower differentiation of the early- and late-flowering lines.InArabidopsis,HOS1modulated the timing ofCONSTANS(CO) accumulation.It is essential to maintain low levels ofFTexpression during the first part of the day and correct photoperiodic response(Lazaroet al.2012).It was predicted that the molecular mechanism and regulatory pathway underlying flowering time in zucchini were different fromArabidopsis.The expression ofCpSOC1was positively correlated with early flowering, which was speculated that the expression level ofCpSOC1was positively correlated with early flowering, and flowering time may be regulated by the ambient temperature, not the other signaling pathways in zucchini.The early flowering functions and regulatory pathways ofCp4.1LG20g08050need to be further validated by performing more molecular biological experiments in zucchini.
5.Conclusion
Early flowering promotes early maturity and production and the capacity to counteract biotic and abiotic stresses; thus,it is an important agronomic trait in zucchini.Our study revealed that zucchini inbred line ‘19’ showed stable early flowering, taking significantly fewer DFF than inbred line‘113’.Genetic analysis revealed that DFF is an inheritable quantitative trait controlled by multiple genes.Based on QTL-seq combined with linkage analysis and QTL mapping of DFF with InDel markers.one major locus was identified under three environmental conditions.It was located in a 117-kb candidate region on chromosome 20.Based on gene annotation, gene sequence alignment, and qRT-PCR analysis, we found that theCp4.1LG20g08050gene may be a candidate gene for the opposite regulation of earliness in zucchini.These results lay a foundation for understanding early flowering better and improving early flowering-based breeding strategies in zucchini.
Acknowledgements
This research was supported by the grants from the National Natural Science Foundation of China (32072590 and 32002051), the China Postdoctoral Science Foundation(2019M661244), and the Academic Backbone Foundation of Northeast Agricultural University, China (20XG03).
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendicesassociated with this paper are available on https://doi.org/10.1016/j.jia.2022.09.009
 Journal of Integrative Agriculture2023年11期
Journal of Integrative Agriculture2023年11期
- Journal of Integrative Agriculture的其它文章
- Germplasm and molecular breeding in horticultural crops
- Development and application of KASP marker for high throughput detection of the seedless trait in grapevine
- A novel mutation in ACS11 leads to androecy in cucumber
- Comprehensive analysis of the full-length transcripts and alternative splicing involved in clubroot resistance in Chinese cabbage
- Virucidal activity of MICRO-CHEM PLUS against African swine fever virus
- Effects of the combined application of organic and chemical nitrogen fertilizer on soil aggregate carbon and nitrogen: A 30-year study