
Comprehensive analysis of the full-length transcripts and alternative splicing involved in clubroot resistance in Chinese cabbage
2023-11-18 09:34:40SUHenanYUANYuxiangYANGShuangjuanWElXiaochunZHAOYanyanWANGZhiyongQlNLiuyueYANGZhiyuanNlULiujingLlLinZHANGXiaowei
Journal of Integrative Agriculture 2023年11期
SU He-nan, YUAN Yu-xiang#, YANG Shuang-juan, WEl Xiao-chun, ZHAO Yan-yan, WANG Zhi-yong, QlN Liu-yue, YANG Zhi-yuan, NlU Liu-jing, Ll Lin, ZHANG Xiao-wei#
1 Institute of Horticulture, Henan Academy of Agricultural Sciences, Zhengzhou 450002, P.R.China
2 School of Materials Science and Engineering, Central South University, Changsha 410083, P.R.China
Abstract Chinese cabbage is an economically important Brassica vegetable worldwide, and clubroot, which is caused by the soilborne protist plant pathogen Plasmodiophora brassicae is regarded as a destructive disease to Brassica crops.Previous studies on the gene transcripts related to Chinese cabbage resistance to clubroot mainly employed RNA-seq technology,although it cannot provide accurate transcript assembly and structural information.In this study, PacBio RS II SMRT sequencing was used to generate full-length transcriptomes of mixed roots at 0, 2, 5, 8, 13, and 22 days after P.brassicae infection in the clubroot-resistant line DH40R.Overall, 39 376 high-quality isoforms and 26 270 open reading frames(ORFs) were identified from the SMRT sequencing data.Additionally, 426 annotated long noncoding RNAs (lncRNAs),56 transcription factor (TF) families, 1 883 genes with poly(A) sites and 1 691 alternative splicing (AS) events were identified.Furthermore, 1 201 of the genes had at least one AS event in DH40R.A comparison with RNA-seq data revealed six differentially expressed AS genes (one for disease resistance and five for defensive response) that are potentially involved in P.brassicae resistance.The results of this study provide valuable resources for basic research on clubroot resistance in Chinese cabbage.
Keywords: Chinese cabbage, clubroot, full-length transcriptome, SMRT sequencing, alternative splicing
1.lntroduction
Clubroot is considered a devastating disease toBrassicacrops and is caused by the soil-borne protist plant pathogenPlasmodiophorabrassicae(Dixon 2009).In recent years, an increasing number of studies have focused on dynamic changes in gene expression to explore the molecular mechanisms of clubroot resistance inBrassicaspecies aided by RNA-seq (Chenet al.2016;Galindo-Gonzálezet al.2020).InBrassicarapa, Chuet al.(2014) used RNA sequencing to segregate clubroot resistant and susceptible populations inoculated withP.brassicae.The results showed that the genes involved in jasmonate and ethylene signalling and the defensive deposition of callose were significantly upregulated in plants carryingRcr1at 15 days after infection, a time when the infection of the root cortex was initiated but clubbing symptoms were not yet visible (Chuet al.2014).Moreover, the identified differentially expressed genes (DEGs) are related to hormone signalling, cell wall modification, NBS-LRR proteins, Ca2+signalling,defense-related callose deposition, chitin metabolism,and pathogenesis related-like protein (PR) in response toP.brassicaeinBrassicacrops (Zhanget al.2016; Ninget al.2019).Illumina RNA-seq is a second-generation sequencing technology that reflects the number and types of genes and provides an important basis for understanding the various expression models of different genes and the regulatory mechanisms of different biological processes in plants (Fuet al.2021; Zhaoet al.2021).However, the accuracy of transcriptome abundance calculations is severely limited by the short length of the sequenced reads obtained and unreliable assembly results (Zhuet al.2020; Niet al.2021).
Single-molecule real-time (SMRT) sequencing technology is a third-generation sequencing technique that generates more uniform coverage, longer read lengths, and high-quality full-length cDNA sequences without post-sequencing assembly compared with RNA-seq (Wuet al.2019, 2020).Therefore, SMRT sequencing can provide an accurate method for novel gene discovery, as well as exon–intron structure and alternative splicing analyses (Donget al.2015).SMRT has been successfully applied to full-length transcriptome analysis in plants in order to obtain highconfidence transcripts (Chaoet al.2018; Maet al.2019).As two significant patterns of posttranscriptional regulation, alternative splicing (AS) and alternative polyadenylation (APA) can greatly improve the diversity level of transcripts (Elkonet al.2013; Wanget al.2021).AS is a crucial posttranscriptional regulatory mechanism that exists widely in plants, and five different types of AS events, including intron retention, exon skipping,alternative 3´ splice sites, alternative 5´ splice sites and mutually exclusive exons, have been shown to play key roles in plant development, growth and stress responses(Staiger and Brown 2013; Mandadi and Scholthof 2015).As an equally important pattern, APA can also create a variety of transcript isoforms with various 3´ ends (Elkonet al.2013).Thus far, few reports have been published on full-length transcriptome sequencing of Chinese cabbage, and the relationships between AS and APA and clubroot resistance have not been revealed.
Chinese cabbage (BrassicarapaL.ssp.pekinensis)is an economically important cruciferous vegetable worldwide.The incidence of cruciferous clubroot disease has increased along with the intensified Chinese cabbage breeding and cultivation recent years, leading to significant losses in Chinese cabbage quality and yield(Yuanet al.2021).Previous to this study, little information was available on posttranscriptional regulation, in particular on AS events, APA events, and other isoforms.In the present research, PacBio RS II SMRT sequencing was applied to generate the full-length transcriptomes of one Chinese cabbage doubled haploid (DH) line (the clubroot-resistant line DH40R) afterP.brassicaeinfection.Then, these transcriptomes were used to predict the open reading frames (ORFs), functions, transcription factors(TFs), long noncoding RNAs (lncRNAs), and AS and APA events.The results of this study will improve the annotation of the current Chinese cabbage genome and provide valuable resources for basic research on clubroot resistance in Chinese cabbage.
2.Materials and methods
2.1.Plant materials and treatments
The Chinese cabbage clubroot-resistant (R) line DH40R used in this work was produced from an isolated microspore culture of the same R individuals from a BC5(Chinese cabbage clubroot-susceptible (S) line Y510-9×European turnip R line ECD01-1)×Y510-9) backcross.DH40R plants showed no gall formation during any of the stages after inoculation (Appendix A).Seeds were germinated on wet filter paper for 3 days and then transferred to plastic trays in which they were maintained at 20–25°C with a photoperiod of 16 h of light and 8 h of darkness at the Institute of Horticulture, Henan Academy of Agricultural Sciences.The pathogen used and the methods for obtainingP.brassicaesuspensions were as described in our previous study (Yuanet al.2021).Individually infected 20-day-old seedlings were selected for full-length transcriptome analysis at 0, 2, 5, 8, 13, and 22 days after inoculation (dai).Sampled roots from five separate plants were combined at 0 and 2 dai.Sampled roots from three individual plants were pooled at 5, 8, 13,and 22 days, and all obtained samples were promptly frozen in liquid nitrogen and stored at –80°C.
2.2.RNA extraction, library preparation and PacBio sequencing
For the subsequent investigations, the total RNA samples from six time points (0, 2, 5, 8, 13, and 22 days) were combined.A typical TRIzol technique was used to isolate the RNA from DH40R cells.Using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, California,USA), the integrity of the RNA was evaluated.Total RNA samples with an RNA integrity number (RIN)≥8 were used to construct the cDNA libraries for PacBio sequencing.Using the Clontech SMARTer PCR cDNA Synthesis Kit(TaKaRa Biotechnology, Dalian, China), 4 μg of RNA was synthesized into cDNA and subsequently amplified to generate double-stranded cDNA.The cDNA was then size selected for fractions <4 kb and >4 kb using the BluePippin™ Size Selection System (Sage Science,Beverly, MA, USA).A SMRTbell library was constructed using 1 μg of size-selected cDNA with the Pacific Biosciences SMRTbell Template Prep Kit.The binding of SMRTbell templates to polymerases was conducted using the Sequel II Binding Kit, and then primer annealing was performed.Sequencing was performed on the Pacific Biosciences Sequel II platform by Annoroad Gene Technology Company (Beijing, China).
2.3.Data processing
First, the smrtlink8.0 package (PacBio) was used to process the sequence data, and a circular consensus read (CCS)was generated with settings of minimum number of full passes=3 and minimum sequence accuracy=0.8.The CCS was then divided into full-length nonchimeric reads and non-full-length reads based on the criteria that the former must contain 5´ and 3´ primer sequences and polyA sequences before the 3´ primer sequences, while the latter must not.The Iso-Seq pipeline contained in the SMRT analysis software package was used to remove poly(A) and chimeric sequences from the above full-length sequences to obtain full-length nonchimeric consistent transcripts,and then cluster and correct errors were identified in order to obtain high-quality and low-quality transcripts.The corrected high-quality transcripts were mapped to theB.rapagenome (http://brassicadb.org/brad/datasets/pub/Genomes/Brassica_rapa/V3.0/) by gmap-2017-08-15, and alignments in the sam format were obtained.The PacBio SMRT reads generated in this study have been submitted to the BioProject database of the National Centre for Biotechnology Information (accession number CNP0002684).
2.4.Prediction of open reading frames (ORFs) and functional annotation of transcripts
The ORFs in transcripts were predicted by the TransDecoder v3.0.1 package (Haaset al.2013).Fulllength transcripts were designated as the transcripts containing complete ORFs and 5´ and 3´-untranslated regions (UTRs).The ORFs were functionally annotated by mapping to the Pfam, COG, Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG), Gene Ontology (GO), NCBI nonredundant protein sequences(NR), NCBI nonredundant nucleotide sequences (NT), and NCBI nonredundant nucleotide sequences (NT) databases using BLAST (version 2.2.28).
2.5.Prediction of long noncoding RNAs (lncRNAs)and transcription factors (TFs)
The lncRNAs were predicted using the Coding-Noncoding Index (CNCI) (Sunet al.2013), the Coding Potential Assessment Tool (CPAT) (Wanget al.2013), and the Coding Potential Calculator (CPC) (Konget al.2007) to predict the coding potentials of transcripts.The Plant Transcription Factor Database (PlantTFDB) was used to analyze the TFs.
2.6.Alternative splicing and alternative polyadenylation analysis
AS events, including retained intron (RI), alternative 5´ splice site (A5SS), alternative 3´ splice site (A3SS),skipped exon (SE), and mutually exclusive exon (MXE),were identified by the Astalavista tool using the default parameters for known and new transcripts (Foissac and Sammeth 2007).APA sites were identified using TAPIS(Abdel-Ghanyet al.2016).
2.7.Alternative splicing validation
To verify the AS events detected with PacBio sequencing,RT-PCR was performed for three randomly selected genes,BraA09g059520.3C,BraA09g043410.3CandBraA01g016250.3C.Total RNA was extracted from the six DH40R samples (0, 2, 5, 8, 13, and 22 dai) using a TaKaRa MiniBEST Plant RNA Extraction Kit (TaKaRa,Dalian, Liaoning, China).The RNA was reverse transcribed into cDNA by a RevertAid First Strand cDNA Synthesis Kit (TaKaRa, Dalian, Liaoning, China).The cDNA was amplified using primers designed with PRIMER PREMIER software (v.3.0) (Appendix B).The PCR products were analyzed following electrophoresis in a 2%agarose gel (Wanget al.2021).
2.8.Expression analysis of AS genes using RNAseq data
To assay the AS gene expression profiles, Illumina RNAseq data of DH40R at 0, 2, 5, 8, 13, and 22 dai were downloaded from NCBI (PRJNA692311).
2.9.Weighted correlation network analysis (WGCNA)
Weighted correlation networks were constructed to identify the specific genes that are highly correlated with clubroot resistance.The co-expression network was analyzed using the WGCNA version package in R software.WGCNAs are groups of highly interconnected genes that exhibit similar expression changes associated with specific physiological processes (Langfelder and Horvath 2008).The interaction network of hub genes in a module was visualized using Cytoscape 3.8.0 (Yuanet al.2021).
3.Results
3.1.General properties of SMRT sequencing
To avoid the limitations of the Illumina HiSeq platform,the full-length transcriptome of Chinese cabbage was sequenced using the PacBio Sequel platform.The roots of DH40R lines at 0, 2, 5, 8, 13, and 22 dai were used for fulllength transcriptome sequencing in the present study.Total RNA samples from the six time points (0, 2, 5, 8, 13, and 22 dai) were mixed, and one SMRT library for the DH40R samples was constructed.The sequences that met the most permissive criteria (minimum number of full passes=3 and minimum sequence accuracy=0.8) were then filtered.In total, 345 292 reads of insert (ROIs) were collected from DH40R.After iterative clustering and error (ICE) correction using SMRT analysis software, a total of 315 929 consensus isoforms were identified.Among them, 39 376 consensus isoforms were high-quality (HQ) isoforms (accuracy>99%).The statistical results are shown in Table 1.
3.2.Predictions and functional annotation of the open reading frame
In total, 26 270 ORFs of the clubroot-resistant line DH40R were predicted using TransDecoder.The number, size,and length distributions of ORFs are shown in Fig.1.In DH40R, ORFs with lengths of 400–600 bp accounted for 14.96% (3 930) of all ORFs, followed by those with lengths of 600–800 bp (3 315; 12.62%), 800–1 000 bp(3 037; 11.56%), and 1 000–1 200 bp (2 770; 10.54%).In addition, ORFs with lengths above 3 000 bp accounted for 4.06% (1 069).
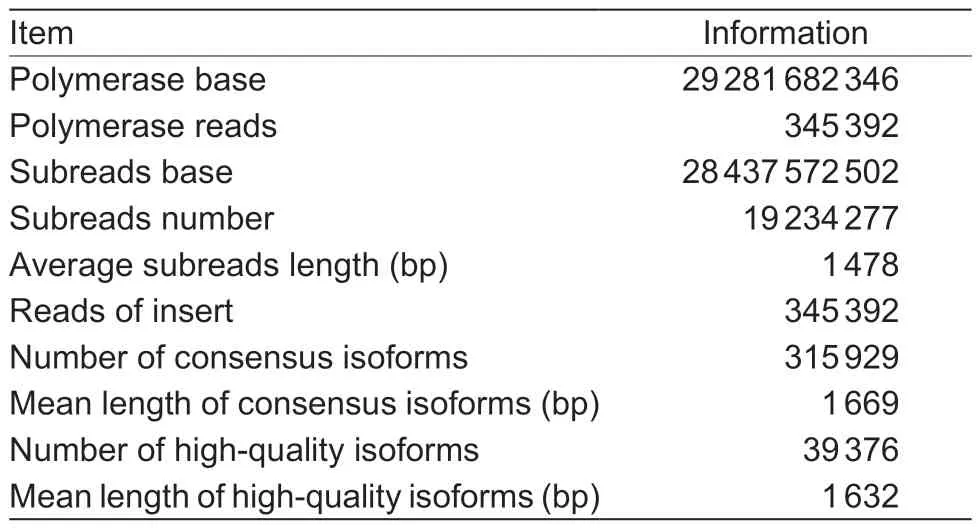
Table 1 The PacBio SMRT sequencing information in the Chinese cabbage doubled haploid clubroot-resistant line(DH40R)
All the ORFs of DH40R were utilized for functional annotation by searching the NR, NT, UniProt, GO,KEGG, and eggNOG databases.The GO analysis results showed that cell processes accounted for the largest proportion, followed by metabolic processes and single-organism processes in biological processes.In the “cellular components” category, the genes involved in cell parts, organelles, membranes, organelle parts,and membrane parts were the most enriched.In the“molecular function” category, binding, catalytic activity and transporter activity were identified as the most enriched (Appendix C).
The KEGG analysis results showed that genes were enriched in pathways related to “cellular processes”,“environmental information processing”, “genetic information processing”, “metabolism”, and “organismal systems” (Fig.2).In the “cellular processes” category,402 genes were involved in transport and catabolism.In the “environmental information processing” category, 700 genes were involved in signal transduction.In the “genetic information processing” category, 706 genes were involved in translation.The ‘‘metabolism’ category mainly consisted of genes involved in carbohydrate metabolism and energy metabolism, and 187 genes were involved in environmental adaptation in the ‘‘organismal systems’’ category.
3.3.Predictions of lncRNAs
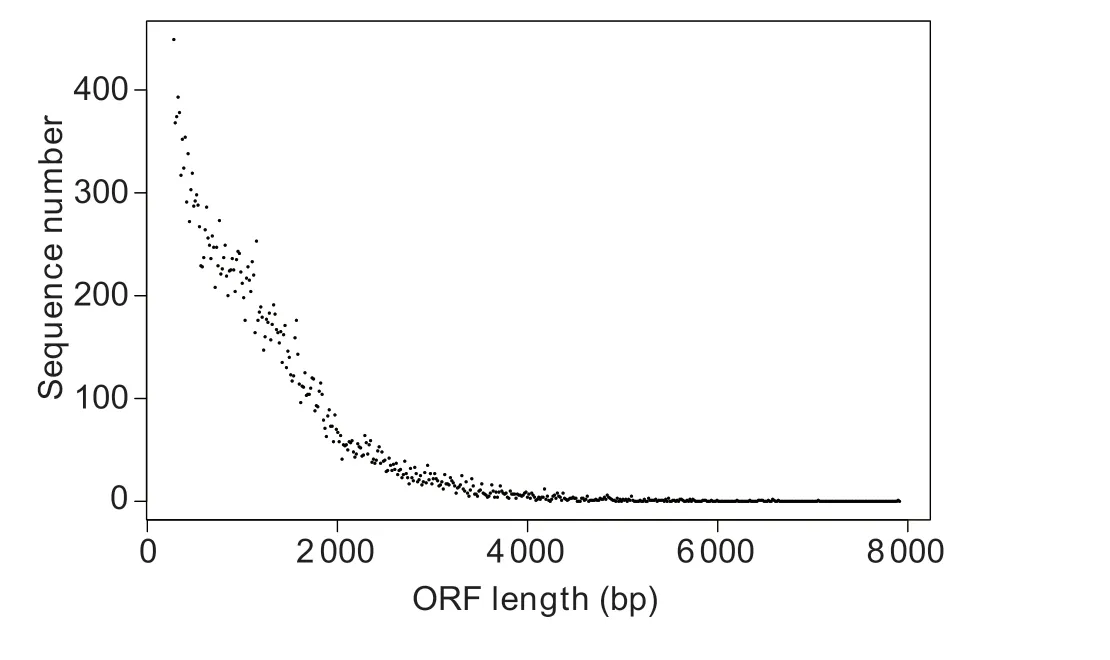
Fig.1 Length distribution of open reading frames (ORF) in the Chinese cabbage doubled haploid clubroot-resistant line(DH40R).
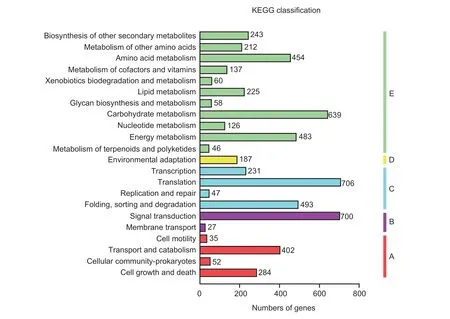
Fig.2 KEGG pathway assignments of genes.A, cellular processes.B, environmental information processing.C, genetic information processing.D, organismal systems.E, metabolism.
LncRNAs are a class of transcriptional RNAs with lengths of more than 200 nucleotides but no proteincoding potential.We used the CPC, CNCI, and CPAT to identify lncRNAs.Based on the results, in the clubroot-resistant line DH40R, the CNCI tool identified 317 lncRNAs, CPC identified 333 lncRNAs, and CPAT identified 331 lncRNAs, with a total of 426 unique lncRNAs identified by all three tools combined (Fig.3-A).The lengths of the lncRNAs varied from 449 bp to 7 959 bp,with the majority of lncRNAs having a length of ≤2 000 bp in DH40R (Fig.3-B).
3.4.ldentification of TFs analysis
TFs are protein molecules that can specifically bind to enhancers and silencers to ensure that the target gene is expressed at a specific time and location with a specific intensity.Here, we identified 56 TF families by comparing the transcript sequences obtained to the sequences in the PlantTFDB 2.0 database (Fig.4).The bHLH (984), MYBrelated (709), NAC (656), WRKY (640), B3 (582), FAR1(580), ERF (514), bZIP (486), C3H (463), and C2H2 (417)were the top 10 TF families enriched in DH40R.
3.5.Analysis of AS and alternative polyadenylation(APA)
AS is an important mechanism of gene expression regulation and proteome diversification.To analyze the AS events in Chinese cabbage, we used AStalavista to characterize splicing variants in the clubroot-resistant line DH40R based on isoform-sequencing (Iso-Seq) data.AS events mostly include the following five types: SEs,A5SSs, A3SSs, MXEs, and RIs.In this study, a total of 1 691 AS events occurred in DH40R (Appendix D).The distribution of all AS types is shown in Fig.5-A.Among the five AS types, MXEs did not occur, while RIs occurred most frequently, accounting for more than 70% of the AS events.To verify the accuracy of the AS events detected with Iso-Seq data, the primers of three randomly selected genes in the DH40R crossing AS region,BraA09g059520.3C,BraA09g043410.3CandBraA01g016250.3C, were designed, and reverse transcription PCR (RT-PCR) was performed.The AS events of those selected genes were visible upon gel electrophoresis, and at least one AS event for each gene is shown in the gel electropherogram in Fig.5-B.
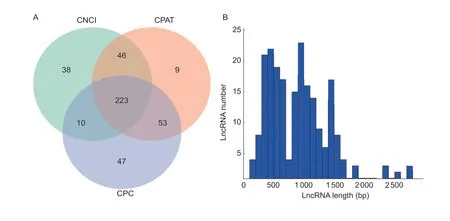
Fig.3 Candidate long noncoding RNAs identified using the Coding-Noncoding Index (CNCI), Coding Potential Assessment Tool(CPAT), Coding Potential Calculator (CPC) and the length distribution of lncRNAs.A, candidate lncRNAs identified using CNCI,CPAT, and CPC in the Chinese cabbage doubled haploid clubroot-resistant line (DH40R).B, length distribution of lncRNAs in DH40R.
In addition to AS, APA can also improve the diversity of the transcriptome by producing transcript isoforms with different 3´ ends.In this study, a total of 1 883 genes with poly(A) sites were identified in DH40R, as shown in Fig.5-C.Among them, 11 genes were found to have at least three poly(A) sites in DH40R.
3.6.ldentification of differential AS genes in response to clubroot
In this study, a total of 1 201 genes were found to have at least one AS event in the clubroot-resistant line DH40R of Chinese cabbage.To investigate Chinese cabbage differential splicing variants in response to clubroot, RNAseq data (Yuanet al.2021) of Chinese cabbage DH40R plants that were inoculated withP.brassicaewere used to analyze AS events.Among all of the 1 201 AS genes, a total of 420 differential AS genes among DH40R-2 daivs.DH40R-0 dai, DH40R-5 daivs.DH40R-2 dai, DH40R-8 daivs.DH40R-5 dai, DH40R-13 daivs.DH40R-8 dai and DH40R-22 daivs.DH40R-13 dai were found in the clubroot-resistant line DH40R after inoculation withP.brassicae(Fig.6; Appendix E).Among these 420 differential AS genes, three genes were shared in DH40R-2 daivs.DH40R-0 dai, DH40R-5 daivs.DH40R-2 dai, DH40R-8 daivs.DH40R-5 dai, DH40R-13 daivs.DH40R-8 dai and DH40R-22 daivs.DH40R-13 dai in the clubroot-resistant line DH40R.Furthermore, 15,44, 47, 33 and 54 differential AS genes were specific in DH40R-2 daivs.DH40R-0 dai, DH40R-5 daivs.DH40R-2 dai, DH40R-8 daivs.DH40R-5 dai, DH40R-13 daivs.DH40R-8 dai and DH40R-22 daivs.DH40R-13 dai,respectively.
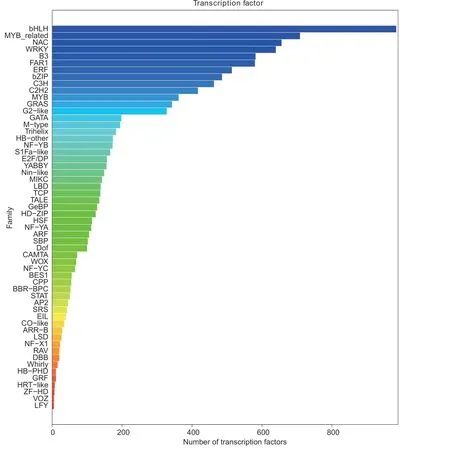
Fig.4 Transcript family distribution of the transcription factors in the Chinese cabbage doubled haploid clubroot-resistant line (DH40R).
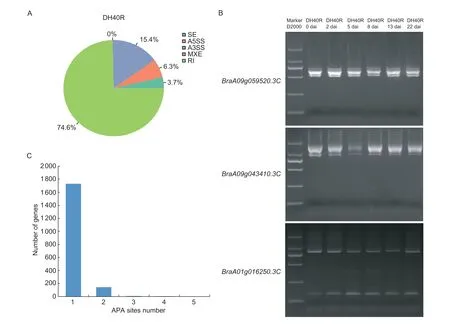
Fig.5 Characteristics of alternative splicing events and alternative polyadenylation sites in the Chinese cabbage doubled haploid clubroot-resistant line (DH40R).A, the distribution of alternative splicing (AS) event numbers.SE, skipped exon; A5SS, alternative 5´ splice site; A3SS, alternative 3´ splice site; MXE, mutually exclusive exon; RI, retained intron.B, the gel electropherograms show the AS events of BraA09g059520.3C, BraA09g043410.3C, and BraA01g016250.3C in DH40R at 0, 2, 5, 8, 13, and 22 days after inoculation (dai) with Plasmodiophora brassicae.C, statistics of alternative polyadenylation sites in DH40R.APA, alternative polyadenylation.
Next, the WGCNA was used to perform a signed weighted gene co-expression network analysis of the 420 differential AS genes (Fig.7-A and B).The turquoise module, with 64 identified genes, was highly associated with DH40R at 8 dai, which was an important time point since the cortical infection rate of DH40R remained basically unchanged from 8 dai onward in our previous study (Yuanet al.2021).In addition, WGCNA was used to construct gene networks.A total of 23 genes were highlighted after WGCNA and interaction network analyses (Fig.7-C), and these genes were highly expressed at 8 dai (Fig.7-D).In addition,BraA02g025510.3C(probable WRKY transcription factor 40, WRKY40),BraA02g025540.3C(cinnamoyl-CoA reductase 2,CCR2),BraA06g025400.3C(protein BONZAI1,BON1),BraA06g040100.3C(lipid phosphate phosphatase 1,LPP1),BraA03g042180.3C(12-oxophytodienoate reductase 3,OPR3), andBraA07g042230.3C(putative disease resistance protein At4g11170) were involved in the defense response.
4.Discussion
Previous study has widely used short-read nextgeneration sequencing (NGS) technology in plant omics research to explain gene expression levels during plant growth and development and stress response (Liuet al.2020; Huet al.2021).However, the short reads generated by NGS have limitations for accurate transcript assembly, while the SMRT sequencing platform generates full-length isoforms that can effectively resolve the entire transcriptome at the isoform level (Wanget al.2021).Although a previous transcriptome analysis of Chinese cabbage during different stages ofP.brassicaeinfection was conducted by our lab (Yuanet al.2021), most of the generated genes did not represent the full-length cDNA sequences and the results did not provide sufficient gene expression profile information.In this work, the thirdgeneration sequencing technology of SMRT sequencing was used to produce the full-length isoforms of Chinese cabbage varieties (clubroot-resistant DH40R).A total of 315 929 consensus isoforms were identified in DH40R,and 39 376 consensus isoforms were high-quality (HQ)isoforms.

Fig.6 UpSet diagram of AS genes among the comparisons of DH40R-2 dai (R2) vs. DH40R-0 dai (R0), DH40R-5 dai (R5) vs.DH40R-2 dai (R2), DH40R-22 dai (R22) vs. DH40R-13 dai (R13), DH40R-13 dai (R13) vs. DH40R-8 dai (R8) and DH40R-8 dai(R8) vs. DH40R-5 dai (R5).
The key regulatory roles of plant lncRNAs in developmental and differentiation processes as well as responses to abiotic and biotic stresses (Liuet al.2015;Wanget al.2018; Yanget al.2019) have been reported,and at least 10 000 lncRNAs have been recently reported inArabidopsis, rice, wheat, maize and soybean (Chenet al.2019).However, previous studies on the function of lncRNAs have been limited due to several difficulties and challenges.In this study, 426 lncRNAs were predicted using three approaches in clubroot-resistant DH40R, and their functional responses toP.brassicaeinfection in Chinese cabbage warrant further exploration.
TFs can regulate the expression of downstream plant stress response genes directly, and some TF families,including the AP2-ERF, NAC, bHLH, WRKY, bZIP, HSF and MYB families, generally play a key role in the plant stress response and secondary metabolism (Endtet al.2002; Huanget al.2012; Tsuda and Somssich 2015).Our analysis identified TFs from 56 different families, and most transcripts in DH40R belonged to the bHLH, MYBrelated, NAC, and WRKY families.InArabidopsis, the roles of most bHLH and MYB-related TFs, including the regulation of plant morphogenesis, flavonoid biosynthesis,hormone signalling and stress responses, have been functionally characterized (Felleret al.2011).NAC TFs are implicated in diverse processes, including various plant developmental programs, senescence formation of secondary walls, and stress responses (Olsenet al.2005).WRKY TFs are a large family of regulatory proteins in plants, and they are notably involved in coping with diverse biotic and abiotic stresses (Eulgem and Somssich 2007).These TFs participate in many different aspects of plant growth and stress responses.
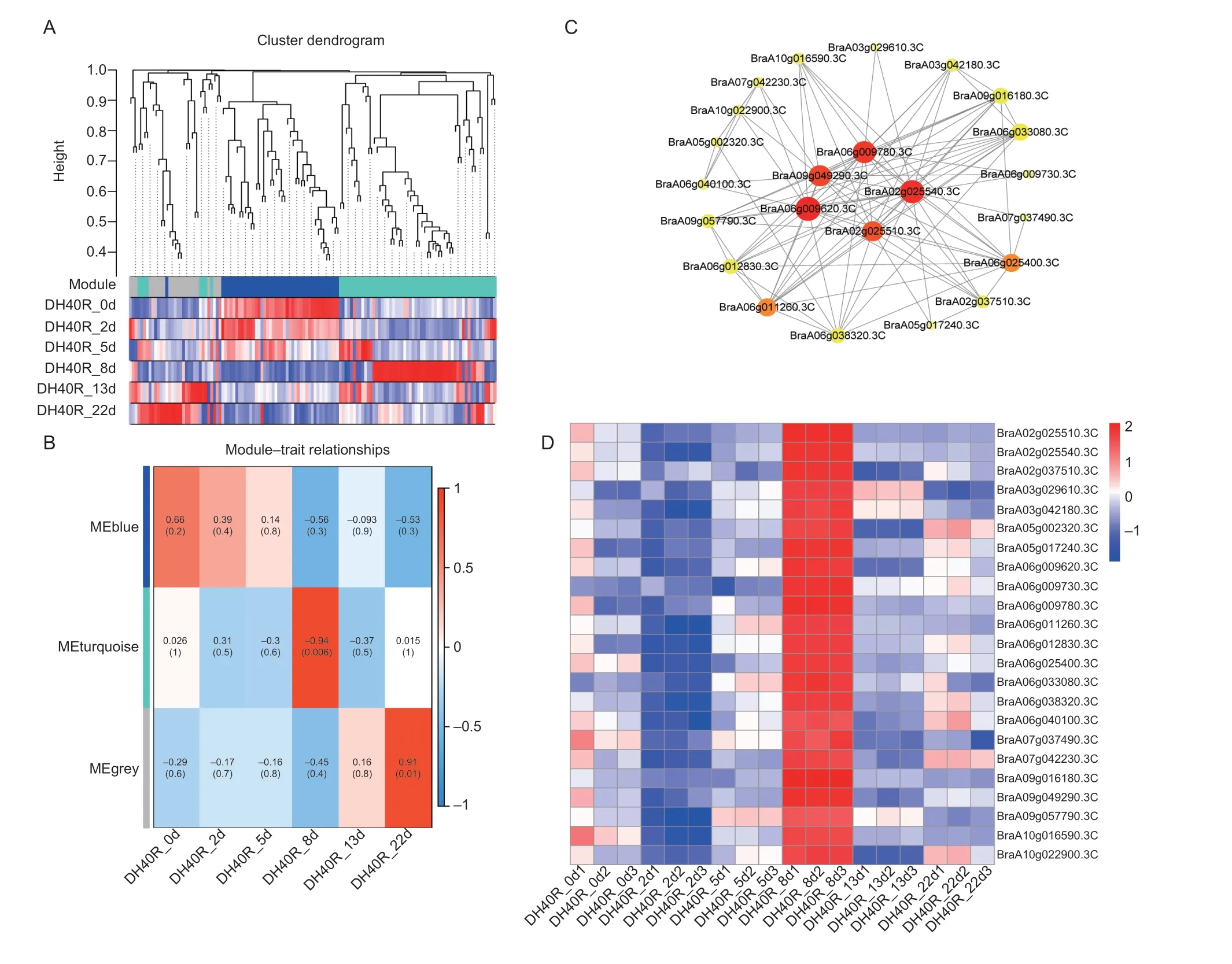
Fig.7 Weighted gene co-expression network analysis (WGCNA) and co-expression network analysis of alternative splicing (AS)genes in DH40R.A, Hierarchical cluster tree showing co-expression modules identified by WGCNA.Each leaf in the tree represents one gene.B, module-sample group association analysis.Each row corresponds to a module, labeled with a color as in A, and each column corresponds to a sample group.C, the correlation networks in the turquoise module.D, the heatmap of 23 genes.
In this work, whole-genome APA analysis was also performed using SMRT sequencing data.From DH40R,1 883 genes were found to have poly(A) sites, of these, at least three poly(A) sites correspond to 11 different genes.These results will provide a basis for further understanding of posttranscriptional regulatory mechanisms in Chinese cabbage.In addition, we detected a total of 1 691 AS events in DH40R, accounting for more than 70% of RI events.These results indicate that full-length reads generated from SMRT sequencing can aid in distinguishing AS events without sequencing assembly.
As the main source of structural and functional polymorphisms of transcripts and proteins, a transcriptional regulation mechanism that can address the infection of many pathogens in plants is important (Howardet al.2013;Songet al.2017; Zhenget al.2017; Zhuet al.2018).AS has been identified as a key player in the regulation of plant defence against pathogen infections (Zhang and Gassmann 2007; Tanget al.2013).InArabidopsisthaliana, the exons increased by more than 44% in plants after inoculation withPseudomonassyringae, which suggests that AS is induced in plants by the pathogen.In addition, severalRgenes need alternately spliced transcripts to produce R proteins that specifically recognize pathogen invasion (Yanget al.2014).Transgenic plants containing only the full-length transcript do not display autoimmunity or the lesion mimic phenotypes induced by increased R protein activity, which provides evidence that AS is unlikely to negatively regulateRgene function.Furthermore, overexpression of the GRP7 protein leads to increased resistance toPseudomonassyringaeand regulates differentPRtranscripts related to salicylic acid and jasmonic acid transcript expression (Streitneret al.2012; Hackmannet al.2014).
In this study, a total of 1 201 genes were shown to have at least one AS event that was found to be induced byP.brassicae.Specifically, compared with our previous RNA-seq data, 420 differential AS genes were found,and six differential AS genes were highlighted after the WGCNA and interaction network analyses, including one disease resistance protein (BraA07g042230.3C, putative disease resistance protein At4g11170) and five defence response genes (BraA02g025510.3C, probable WRKY transcription factor 40;BraA02g025540.3C, cinnamoyl-CoA reductase 2;BraA06g025400.3C, protein BONZAI1;BraA06g040100.3C, lipid phosphate phosphatase 1; andBraA03g042180.3C, 12-oxophytodienoate reductase 3).The geneAt4g11170refers to the disease-resistance geneRMG1(Resistance Methylated Gene 1), and it consists of a complex of TIR-NB-LRR genes for the defence response (Meyerset al.2003).The expression ofWRKYgenes in plants is involved in the defence against phytopathogens.WRKY transcription factor 40 is involved in the transcriptional regulation of defencerelated genes in response to fungal pathogens and hormonal stimuli in canola (BrassicanapusL.) (Yanget al.2009).Lignification is one plant defence response to pathogen attack, and cinnamoyl-CoA reductase (CCR)plays a role in the biosynthesis of lignins.In response toXanthomonasinfection,AtCCR2plays a key role in the establishment of disease resistance mechanisms inArabidopsisthaliana(Lauvergeatet al.2001).As osmotic stress signalling components, the copine proteins BONZAIs (BON) regulate stress responses, including early Ca2+signalling, gene expression reprogramming,ABA accumulation, and plant growth, while BON1 plays a major role in controlling Ca2+signalling, SA accumulation and plant growth in soil without osmotic stresses (Chenet al.2020).AtLPP1knockout mutant plants were used to determine the roles ofAtLPP1in lipid metabolism and cellular responses to stress, and the results showed that theAtLPP1-encoded lipid phosphate phosphatase enzyme may attenuate the signalling functions of phosphatidate and/or diacylglycerol pyrophosphate (Pierrugueset al.2001).12-Oxophytodienoic acid (OPDA) is a signaling molecule that regulates defence and stress responses in plants, and 12-oxophytodienoate reductase (OPR) is involved in the biosynthesis of jasmonic acid and triggers the conversion of OPDA into OPC-8:0 (Huanget al.2022).The results of this study suggest that AS may contribute to the adjustment to pathogen invasion in Chinese cabbage,and that the selected differential AS genes may be involved in the regulation of resistance toP.brassicae.
5.Conclusion
PacBio SMRT sequencing was applied to generate the full-length transcriptome of mixed roots in the clubrootresistant line DH40R afterP.brassicaeinfection.The number and mean length of the isoforms, as well as the number of complete ORFs, were determined from the SMRT sequencing data; in addition, lncRNAs and transcription factors were annotated.Furthermore, 420 differential AS genes were discovered in the clubrootresistant line DH40R when compared to our earlier RNAseq data, and six differential AS genes, including one disease-resistance protein and five defense response genes, may be regulated by resistance toP.brassicae.The findings of this study can facilitate further studies on the clubroot resistance mechanism in Chinese cabbage.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31872945 and 31801874), the earmarked fund for China Agricultural Research System (CARS-23-G15), the Funds for Distinguished Young Scientists from Henan Academy of Agricultural Sciences, China (2021JQ03), and the Innovation Team of Henan Academy of Agricultural Sciences, China (2021TD06).
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendicesassociated with this paper are available on https://doi.org/10.1016/j.jia.2022.09.014
 Journal of Integrative Agriculture2023年11期
Journal of Integrative Agriculture2023年11期
- Journal of Integrative Agriculture的其它文章
- Germplasm and molecular breeding in horticultural crops
- Development and application of KASP marker for high throughput detection of the seedless trait in grapevine
- QTL analysis of early flowering of female flowers in zucchini(Cucurbita pepo L.)
- A novel mutation in ACS11 leads to androecy in cucumber
- Virucidal activity of MICRO-CHEM PLUS against African swine fever virus
- Effects of the combined application of organic and chemical nitrogen fertilizer on soil aggregate carbon and nitrogen: A 30-year study