
Immunoglobulin A vasculitis nephritis: Current understanding of pathogenesis and treatment
2023-11-16 05:47MichelaAmatrudaNicolinaStefaniaCarucciRobertoChimenzGiovanniConti
World Journal of Nephrology 2023年4期
Michela Amatruda, Nicolina Stefania Carucci, Roberto Chimenz, Giovanni Conti
Abstract
Key Words: Immunoglobulin A vasculitis nephritis; Immunoglobulin A vasculitis;Henoch-Schoenlein purpura; Immunoglobulin A nephropathy; Vasculitis, glomerulonephritis
INTRODUCTION
Immunoglobulin A (IgA) vasculitis (IgAV), also known as Henoch-Schoenlein purpura (HSP), is the most common systemic vasculitis in children.It is defined by the presence of non-thrombocytopenic palpable purpura or petechiae(mandatory criterion) predominantly located in the lower limbs, plus abdominal pain or IgA deposition in tissue biopsy or arthritis/arthralgia or renal disease[1,2].20%-80% of patients with IgAV develop renal involvement, termed IgAV nephritis (IgAVN), which is the key element in affecting long-term outcome[3-5].
According to the European SHARE (Single Hub and Access point for paediatric Rheumatology in Europe) plane[6],all IgAV patients should be investigated for renal involvement at diagnosis and throughout follow-up by measuring blood pressure, determining the presence of haematuria, quantifying albuminuria and/or proteinuria and, finally, estimating glomerular filtration rate (eGFR)[7].If initial tests are normal, they need to be monitored for at least 6-12 mo[8-12].The renal biopsy should be performed in the case of impaired eGFR, persistent proteinuria, nephrotic (e.g., severe proteinuria,low serum albumin levels and oedema) or nephritic (e.g., impaired eGFR, hypertension, haematuria/proteinuria)syndrome.
This review summarizes some important current aspects of IgAVN and most importantly the understanding of its pathogenesis and treatment founded on the results of a multitude of clinical and experimental researches.
IGA NEPHROPATHY AND IGAVN
IgA nephropathy (IgAN) is the most common primary glomerulonephritis in children and adults.Exclusive renal involvement is characterized by a very slow progression, typical of chronic diseases[9].In contrast, IgAVN is the most common cause of secondary glomerulonephritis in pediatric age; renal involvement tends to have an acute and selflimiting course similar to that of post-infectious glomerulonephritis, but if persistent it can lead to chronic kidney disease and end-stage renal disease approximately 20 years after diagnosis[10,11].
In a retrospective study based on the epidemiological, clinical and laboratory characteristics of IgAV patients, Carucciet al[12] investigated the initial risk factors for IgAVN development, suggesting that age at diagnosis and abdominal pain were associated with a higher risk of onset of the kidney disease in pediatric age.However, additional long-term studies are still needed for the retrospective study type[12].
According to a Japanese study conducted by Komatsuet al[13], IgAVN has two peaks in incidence (1-19 years and 60-69 years), while IgAN has an isolated peak in incidence during the 4thdecade.Compared to IgAN, IgAVN is clinically more severe (especially considering the parameters of proteinuria and hypoalbuminemia), and histologically more aggressive, with a higher percentage of proliferative endocapillary glomerulonephritis and crescentic glomerulonephritis[13].
Despite these differences, several studies have shown that the two diseases share the same pathogenesis, namely a defect in IgA1 glycosylation[14-16].In addition, glomerular histologic findings can be identical, ranging from focal proliferative lesions with diffuse mesangial IgA deposits to extracapillary proliferative lesions with crescent formation, making it sometimes impossible to distinguish IgAN from IgAVN in the absence of extrarenal signs.For this reason, the two entities have recently been considered to be two different manifestations of the same pathology and in 2012, during the Chapel Hill Consensus Conference on the nomenclature of vasculitis, it was decided to replace the definition "HSP" with that of "IgAV", considering it the systemic form of IgAN[17,18].
In an interesting case series by Kameiet al[10], 11% of patients with an initial diagnosis of IgAN subsequently (over a period ranging from 5 mo to 14 years) developed palpable purpura, which allowed the diagnosis to be changed to IgAVN, supporting the hypothesis that IgAN and IgAVN are actually two variants of the same disease.In almost all of these patients, worsening of nephritis was described after the onset of purpura[10].For this reason, patients diagnosed with IgAN require careful and prolonged follow-up with special attention to the possible appearance of purpura, a sometimes nuanced finding that therefore risks going unnoticed[19].
PATHOGENESIS
IgAVN shares many pathophysiological features with IgAN (Table 1).The aim of this review is to update the pathogenesis of IgAVN by providing more information regarding impaired glycosylation of IgA1, generation of immune complexes and their kidney deposition, and finally concerning complement activation and stimulation of the mesangial cells, which leads to their expansion and cytokine release[20].Toll-like-receptor (TLR) activation and B cell proliferation,infection triggers, and genetic factors are also involved in the IgAVN pathogenesis[21].
Infection triggers
Helicobacter pylori[22],Streptococcus pneumoniaeandHaemophilus Influenzae[23,24] are the main pathogens associated with the disease.Infections may be involved in the pathogenesis of IgAVN through two mechanisms.The first is due to the presence of N-acetylgalactosamine (GaINAc) on the superficial side of pathogens stimulating the output of cross-reactive IgA and IgG that identify galactose-deficient IgA1 (Gd-IgA1).Alternatively, microbial agents containing antigens similar to structures on the vessel wall generate a cross-reactive autoantibody response[25].

Table 1 Mechanism of immunoglobulin A vasculitis nephritis pathogenesis
Genetic factors
Little is known yet about the role of genetic factors in the IgAVN pathogenesis, although the presence of ethnic and geographical differences in the incidence of IgAV and IgAVN would indicate its involvement[26,27].Furthermore,several genes involved in cytokine and chemokine production, complement activation, and regulation of endothelium activity have been implicated in IgAV susceptibility[25,28].
Serum levels of Gd-IgA1 are heritable in both IgAN and IgAVN, indicating that the genetic predisposition to develop IgAVN and IgAN is the same[16].
Impaired glycosylation of IgA1
IgAVN and IgAN appear to have identical pathophysiology, with only quantitative differences[29].High levels of IgA and IgA-containing immune complexes have been observed in both cases.However, this serum abnormality is not a sensitive marker for diagnosis[30].IgA1, not IgA2, is the main component of IgAN and IgAVN.The IgA1 molecule has a hinge region which contains up to six O-linked glycan chains made up GaINac, typically with β1,3-linked galactose (Gal)attached to it[31,32].Normally, mono- and di-sialylated Gal-GaINAc disaccharides are present in healthy subjects[33,34].Elevated levels of Gd-IgA1 have been detected in patients with IgAVN and IgAN, compared with the healthy population and patients with other glomerular diseases[35].Therefore, Gd-IgA1 now plays a pivotal role in both IgAN and IgAVN pathogenesis[36].
It has been reported that these patients have decreased galactosylation of O-glycans, resulting in reduced β1,3-galactosyltransferase activity in the peripheral B cells of patients.Reduced β1,3-galactosyltransferase activity leads to a lack of terminal β1,3-galactosyl residues in the hinge region of IgA1[15,37].
Gd-IgA1 polimer molecules are known to be anti-glycan IgA1 or IgG, resulting in the formation of circulating immune complexes that are deposited in the renal mesangium and subepithelial and subendothelial space, and which incite glomerular injury.In IgAVN, IgA deposit can be found not just in the kidney, but also in other sides like the skin, and larger immune complexes are found compared to IgAN[15].Deposition of IgA1-containing immune complexes appears to be mediated by a relationship with mesangial transferrin 1 (CD71) and CD89 receptors, also known as FcαRI, which may occur as a transmembrane receptor in myeloid cells.These receptors on the mesangial surface are more highly expressed than in healthy children.
The monomeric IgA can bind, but without crossbinding with FcαRI, leading to anti-inflammatory reactions[38].Monovalent FcαRI targeting results in the formation of intracellular structures called "inhibisomes", which obstruct the signaling of nearby activated receptors.This process is referred to as ITAM inhibitory signaling (ITAMi) and results in a downregulation of immune activation.In contrast, IgA immune complexes binding to FcαRI on neutrophils induce activating ITAM signaling, resulting in multiple pro-inflammatory functions.In addition, the activation of FcαRI induces the LTB4 chemoattractant release, resulting in neutrophil migration[39,40].
Consequently, after deposition the mesangial cells start to proliferate and produce other components of the extracellular matrix, and also inflammatory and profibrogenic cells such as cytokines and chemokines[35-41].
Complement activation
The activation of the complement system plays an important role in the pathophysiology of IgAV and IgAVN, including infection triggers and genetics.Indeed, mesangial deposits contain the complement components C3 and C5-C9, which are able to form the attack complex that destroys the membrane of target cells[42].High levels of C3a, C5a and Bb have also been documented in the serum of pediatric patients with acute IgAV.C5a is a neutrophil chemoattractant that increases during systemic inflammation.C3a and C5a increase interleukin (IL)-8 secretion by endothelial cells, further attracting neutrophils.
More recently, the lectin-related complement activation pathway has also been shown to be involved in IgAVN and IgAN, as IgA can activate mannan-related lectins[43,44].In contrast, IgA cannot activate the classic pathway and the main activator of the classic pathway, C1q, which is not present in immune complexes[45].
TLR activation and B cell proliferation
Another mechanism considered to be responsible for IgAV injury is the hyperreactivity of B and T cells in response to specific antigenic triggers.TLR signaling is the first line of defense against microbial infection.Hyperactivated TLR signaling causes cellular inflammatory infiltration that generates cytokines and autoantibodies, leading to autoimmune diseases[46,47].
TLRs are expressed in numerous cells, including kidney cells.There is increasing evidence to support the role for TLRs in autoimmune and inflammatory kidney diseases[22].
Donadioet al[48] showed children with IgAV that had significantly increased expression of mRNA encoding for TLR4,compared with healthy controls.TLR2 mRNA expression showed a borderline increase, while no difference was found in the expression of mRNA encoding for TLR3 and TLR9.Regulatory T cells (Treg) expressing the transcriptional factor FoxP3 play a potent anti-inflammatory role.Defective expression of FoxP3 mRNA and reduced expression of transforming growth factor-β1 mRNA were also demonstrated, indicating a defective activity of Treg cells in IgAV[48].
However, it is still unclear if TLR expression is also associated with IgAVN development.
The role of anti-endothelial cell antibodies in IgAVN
Anti-endothelial cell antibodies (AECAs) are a heterogeneous group of antibodies directed to human endothelial cell antigens.A role of these antibodies in IgAVN has been hypothesized[49].According to the hypothesis, tumor necrosis factor-α can increase the binding of IgA1 AECAs to endothelial cells.The latter produce IL-8, leading to neutrophil migration[50].
The interaction between IgA1 AECAs and FcαRI on neutrophils results in the release of LTB4, inducing neutrophil recruitment, reactive oxygen species release, neutrophil extracellular traps accompanied by the cell death (NETosis) and antibody-dependent cellular cytotoxicity.All this leads to vascular injury in the final analysis[51].
Other possible pathogenic mechanisms of IgAVN
Other pathogenetic mechanisms underlying IgAVN need to be further investigated.Masudaet al[52] showed that nephritis-associated plasmin receptor, a nephritogenic antigen for acute poststreptococcal glomerulonephritis, might have a pathogenic role in a subgroup of patients with IgAVN[52].
In a study by Davinet al[29], elevated plasma levels of IgE were found more commonly in patients with IgAN, but the pathogenetic role of IgE has not yet been clarified.In fact, mast cells are not usually present in the mesangium[24].
Eosinophil activation, higher levels of eosinophil cationic protein (ECP) in serum, and renal α-smooth muscle (α-SMA)expression have also been proposed as playing a role in the pathogenesis of IgAVN[25].
Observations indicate that enhanced renal α-SMA expression could be an early histological marker of IgAN progression.Similarly, increased expression of α-SMA in the tubule-interstitial region, but not in glomeruli, has been associated with bad prognosis[26].
BIOMARKERS
It is currently not possible to predict and identify which children may develop chronic kidney damage from onset.Some serum and urine markers can identify IgAV patients with or without renal involvement and in predicting the severity of renal inflammation to avoid chronic damage[53,54].Measuring biomarkers in urine has many advantages: Samples are easy to collect using non-invasive methods; urine reflects damages in renal parenchyma, unlike blood, which comes into contact with many organs and organ systems; the number of different core proteins in the urine is lower than in blood[55].In a prospective, multicenter study, Pilleboutet al[54] identified biomarkers that may identify IgAVN at the onset of the disease: Serum Gd-IgA1 level, urine IgA, IgG, IgM, IL-6, IL-8, IL-10, and IgA-IgG and IgA-sCD89 complex levels[54].A systematic literature review performed by Suginoet al[56] identifies that some clinical and urinary biomarkers potentially correlate with the presence and severity of IgAVN in children.The most promising preclinical urinary biomarkers in predicting nephritis are: Kidney injury molecule-1 (KIM-1), monocyte chemotactic protein-1 (MCP-1), Nacetyl-β-glucosaminidase (NAG), and angiotensinogen.Urinary KIM-1, MCP-1, and NAG correlate with the disease severity of nephritis (4).However, none of them prove to be established markers of disease.Further studies are needed to verify whether preclinical markers are better than the currently used ones (24-h urinary protein values, urinary protein:creatinine ratio and urinary albumin concentration)[55].
TREATMENT
Figure 1 illustrates the integration of the pathogenesis diagram with the drug treatment diagram to reveal the supporting mechanism of the drugs.
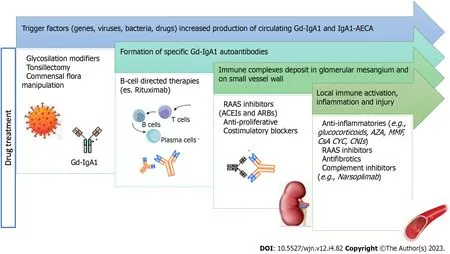
Figure 1 Pathogenesis and drug treatment for immunoglobulin A vasculitis and nephritis.Gd-IgA1: Galactose-deficient-immunoglobulin A; AECA:Anti-endothelial cell antibodies; RAAS: Renin-angiotensin-aldosterone system; ACEI: Angiotensin-converting enzyme inhibitors; ARBs: Angiotensin receptor blockers;AZA: Azathioprine; MMF: Mycophenolate mofetil; CsA: Cyclosporine A; CYC: Cyclophosphamide; CNIs: Calcineurin inhibitors.
Decisions regarding the treatment of IgAVN are challenging due to the large percentage of patients with a positive prognosis and the uncertain clinical progression of single patients.Unfortunately, evidence-based treatment is not yet available even for the most serious event[1].In several retrospective studies, delayed therapy has been related to a bad outcome.Therefore, despite the risk of spontaneous remission, it may be recommended that severely affected patients be treated as soon as possible.
Recently, the Kidney Disease Improving Global Outcomes (KDIGO) guidelines have mainly highlighted that the treatment of IgAVN remains a matter of debate and, in the absence of sufficient long-term data, have recommended that it should be treated in the same way as in patients with non-severe forms of isolated IgAN.However, the KDIGO guidelines do not take into account the more acute onset of IgAVN with more aggressive lesions in renal histology[57].According to KDIGO guidelines, IgAVN patients should first receive a supporting care including routine modification(smoking cessation, weight control, regular exercise and dietary sodium restriction), blood pressure control and a course of renin-angiotensin system (RAS) blockers without immunosuppressive drugs or steroids.In the opinion of experts, this approach can result in the undertreatment of glomerular inflammation, mainly because acute and potentially aggressive glomerular inflammation goes untouched or its immunosuppressed treatment is postponed for several months.
On the contrary, European treatment guidelines consider IgAN and IgAVN as two distinct entities and suggest oral steroids as a first line therapy in IgAVN.In this regard, the German Society of Pediatric Nephrology has recently suggested an early treatment approach for patients with important kidney involvement.Following this treatment suggestion, IgAVN patients with nephritic syndrome, nephrotic syndrome, or glomerular cellular growths will receive an initial standardized corticosteroid- based treatment regimen for 2 mo, followed by an additional immunosuppression in patients with inadequate response after 3-6 mo from the start of treatment[58].
Corticosteroids, intravenous or oral, are part of most treatment regimens, and there is some evidence of their positive result on the long-term outcome of adult IgAN patients.Similarly, other immunosuppressive therapies, such as azathioprine (AZA), mycophenolate mofetil (MMF), cyclosporine A (CsA), or rituximab, have been shown to be effective in individual cases or in small series of patients.Cyclophosphamide (CYC) has also been used for more severe manifestations of IgAN (Table 2)[6].In conclusion, given the rare nature of severe IgAVN, there is a necessity to standardize the diagnostic and therapeutic approach at least nationally, but ideally multinationally, in order to gain more expertise and move to evidence-based treatment.Future treatment strategies should be evaluated in large multicenter trials.Figure 2 summarizes the treatments available for IgA vasculitis and nephritis.
Corticosteroids
Several studies highlight a potential beneficial impact of corticosteroids in IgAVN.Oral prednisolone and/or pulsed methylprednisolone should be used as the earliest treatment in those cases with mild-moderate IgAVN[6].A randomized, placebo-controlled trial showed that IgAVN was resolved more quickly in children treated with prednisone than in those treated with placebo.However, the study offered outcome data only 6 mo after randomization.Hence, it is unknown whether prednisone treatment reduced the number of cases with persistent IgAVN or simply encouraged a more rapid resolution of the renal disease compared with the placebo[59].Kimet al[60] showed that corticosteroid exposure significantly reduced serum Gd-IgA1 levels, which are associated with the pathophysiology of IgAVN[60].
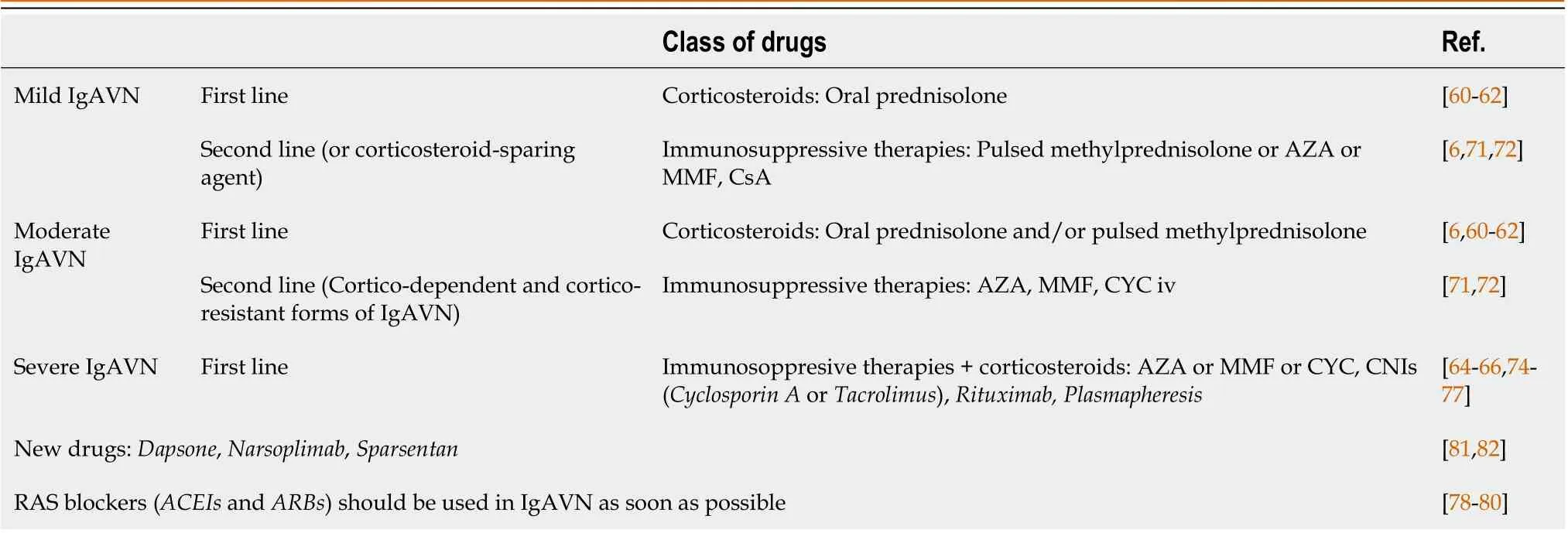
Table 2 Treatments according to European treatment guidelines
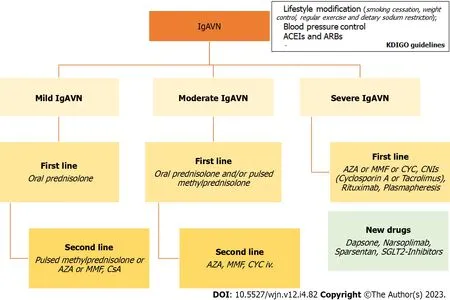
Figure 2 Therapeutic algoritm for immunoglobulin A vasculitis and nephritis.ACEI: Angiotensin-converting enzyme inhibitors; ARBs: Angiotensin receptor blockers; IgAVN: Immunoglobulin A vasculitis nephritis; AZA: Azathioprine; MMF: Mycophenolate mofetil; CYC: Cyclophosphamide; CNIs: Calcineurin inhibitors; CsA: Cyclosporine A.
However, no evidence has been found that early treatment with steroids could prevent nephritis in IgAV patients and reduce the risk of proteinuria in the following 12 mo[61].
Other immunosuppressive therapies
In the cortico-dependent and cortico-resistant forms of IgAVN, other immunosuppressive treatments, such as calcineurin inhibitors (CNIs), AZA, MMF, CYC, rituximab and plasmapheresis can be considered.AZA, MMF or intravenous CYC can be used in the first- or second-line management of moderate-severe IgAVN[6].
CNIs should be considered a hopeful agent for the treatment of severe IgAVN.The two CNIs currently on the market are CsA and tacrolimus.CsA was found to be effective in improving histological lesions and proteinuria in IgAVN patients[62,63].In a pilot study of 20 IgAVN children, 12 of whom received tacrolimus treatment reached complete remission; eight achieved partial remission at the end of 6 mo[64].
Current data are not in favor of CYC use in IgAVN because no statistically significant differences were found in the group of patients treated with this immunosuppressive drug compared to the group treated with high doses of corticosteroid[65,66].CsA or oral CYC cannot be routinely recommended in moderate IgAVN.Intravenous CYC with pulsed methylprednisolone and/or oral prednisolone are also required as an earliest treatment in patients with IgAVN[6].AZA appeared to be an effective steroid-sparing drug.This has allowed all steroid-dependent patients to go steroid-free.No formal guidelines are available for the duration of treatment.In studies no patient has had any adverse events associated with AZA therapy[67,68].
MMF should be suggested patients with IgAVN, especially if proteinuria still remains after an initial steroid course and despite antiproteinuric treatment[69,70].A current meta-analysis has, in fact, reported results combining eight studies and proposed that patients with IgAN treated with MMF had higher remission than the control group[71].In association with steroid therapy, AZA and MMF can be used as a maintenance treatment in those patients with severe IgAVN[6].
Data on the use and efficacy of the anti-CD20 monoclonal antibody rituximab in patients with IgAVN are still limited.A few case reports have recommended that rituximab could be successful in IgAVN in both pediatric and adult ages,especially if other oral immunosuppressive treatments have not been able to induce remission[72,73].
Finally, there is plasmapheresis, which can be used as a rescue therapy in cases of rapid progression to renal failure or persistent nephrotic syndrome.Interestingly, early plasmapheresis has been useful in some patients even without additional immunosuppression[74,75].
RAS blockers
Numerous data suggest that RAS blockers such as angiotensin-converting enzyme inhibitors/angiotensin receptor blockers should be used in IgAVN and as soon as possible.Prospective randomized trials have shown that the use of these drugs improves long-term renal issues in children and adults and prevents/Limits secondary glomerular injury in patients with persistent proteinuria[76-78].
New drugs
Dapsone is a drug that could be considered for the treatment of IgAVN because it can suppress the development of toxicfree radicals by neutrophils.It also reduces the output of IgA antibodies, an essential phenomenon in the pathophysiology of IgAVN[79,80].
A phase II study recently demonstrated the safety, tolerability, and efficacy of narsoplimab, a novel Mannan-binding lectin-associated serine proteinase 2 inhibitor.It is a humanized monoclonal antibody that inhibits the lecithin pathway target of the complement system and seems to result in a clinically significant reduction in proteinuria and the stability of renal function as evaluated by eGFR in high-risk patients with IgAN[81].
A clinical trial is being conducted to evaluate the use of sparsentan as a potential first-line treatment in patients with newly diagnosed IgAN who have not received previous treatment with RAS blockers.Treatment response will be based on endpoints of proteinuria and GFR and will be assessed by changes from the baseline compared to another treatment[76].
Many studies have shown the renoprotective role of sodium-glucose cotransporter 2 (SGLT2) inhibitors in early to advanced diabetic kidney disease.Recent evidence show that SGLT2 inhibitors are similarly renoprotective in nondiabetic chronic kidney disease, such as IgAN, in a wide range of eGFR of 25-75 mL/min/1.73 m2and albumin/creatinine ratio of 200-5000 mg/g[82,83].
INTERVENTIONS FOR PREVENTING IGAVN
According to a recent Cochran study on the prevention and treatment of IgAVN, studies have shown no benefit of prednisone with respect to placebo or no treatment in preventing persistent kidney disease in children without or with little kidney disease at the time of onset[61].
CONCLUSION
According to the European SHARE initiative, only follow-up is required for patients with microscopic haematuria, no renal disorder and proteinuria, or with non-persistent mild or moderate proteinuria.In the case of severe proteinuria or impaired GFR, a paediatric nephrologist must be consulted and a renal biopsy performed.In the case of mild IgAVN, oral prednisolone should be used as a first-line treatment.In some patients with persistent proteinuria, the addition of AZA or MMF, CsA or pulsed methylprednisolone, may be used as a second-line treatment or as a corticosteroid- sparing agent.For patients with moderate IgAVN, oral prednisolone and/or pulsed methylprednisolone should be used as earliest treatment.Addition of AZA, MMF or intravenous CYC may also be used in the first- or second-line treatment of moderate nephritis, according to the histological findings in the kidney biopsy.For severe IgAVN, intravenous CYC with pulsed methylprednisolone and oral prednisolone should be used as a first-line treatment.AZA/MMF plus steroid therapy can be used as a second life[6].
FOOTNOTES
Author contributions:Amatruda M wrote the article and reviewed the references; Carucci NS wrote the article and revised the references;Chimenz R corrected the article; Conti G thought up the article, supervised the chapters of the article and corrected it.
Conflict-of-interest statement:All the authors report no relevant conflicts of interest for this article.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial.See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:Italy
ORCID number:Michela Amatruda 0000-0001-9232-777X; Roberto Chimenz 0000-0001-9143-4637; Giovanni Conti 0000-0002-0617-500X.
S-Editor:Fan JR
L-Editor:A
P-Editor:Zhao S
 World Journal of Nephrology2023年4期
World Journal of Nephrology2023年4期
- World Journal of Nephrology的其它文章
- Moderate stepwise restriction of potassium intake to reduce risk of hyperkalemia in chronic kidney disease: A literature review
- Transcending boundaries: Unleashing the potential of multi-organ point-of-care ultrasound in acute kidney injury
- Role of simulation in kidney stone disease: A systematic review of literature trends in the 26 years