
单原子催化剂在锂硫电池中的应用
2023-11-14 07:56:52张山青
广东工业大学学报 2023年6期
陈 超,雷 源,林 展,2,张山青,2
(1.广东工业大学 轻工化工学院, 广东 广州 510006;2.化学与精细化工广东省实验室 揭阳分中心, 广东 揭阳 515200)
锂硫电池因其超高的理论能量密度 (2 600 Wh·kg-1) 、价格低廉,以及环境友好等特性备受关注,被认为是下一代储能系统的理想材料之一。另一方面,锂硫电池的实际性能受到诸多因素的限制,其中多硫化物(Polysulfides, PSs) 的“穿梭效应”是阻碍锂硫电池商业化的关键问题,也是锂硫电池体系研究的重点及难点[1-3]。早期,研究者们主要采用“吸附”策略应对“穿梭效应”,即利用材料对PSs的亲和力 (吸附作用)锚定PSs并限制其扩散[4-6]。但是,通过简单的吸附作用并不能从根本上解决PSs的扩散问题,因为PSs在材料的表面会很快达到 (吸附) 饱和,限制PSs的进一步吸附。近年来,采用“催化”策略缓解“穿梭效应”得到了广泛关注[7-9]。这种策略旨在通过催化作用提高硫物种之间转化的反应动力学,加快PSs与Li2S2/Li2S之间的转化。这些具有催化作用的极性材料对PSs也有吸附能力,因此可以通过吸附-催化协同作用缓解“穿梭效应”。代表性材料有掺杂杂原子的碳材料[10-11]、过渡金属化合物[12-14]和异质结构材料[15-18]等。
单原子催化剂 (Single-atom Catalysts, SACs) 的概念由张涛等于2011年提出[19],指该类催化剂中催化活性中心 (金属) 以单个金属原子的形式与载体中的杂原子键合并负载于载体表面。由于具有高催化活性位点利用率 (理论上100%的金属原子利用率) 、均匀的金属活性中心、独特的电子结构等特点,SACs在催化领域备受研究者们的青睐。目前,SACs在析氢[20-22]、析氧[23-25]、二氧化碳还原[26-28]等诸多领域均展示出不错的催化性能。
SACs的诸多特性使其在锂硫电池领域中具有应用前景。作为催化剂的活性中心,金属原子对PSs具有强亲和力,可通过吸附作用锚定PSs,并可催化加速硫物种之间的氧化还原反应动力学。催化剂的载体一般为纳米结构、杂原子掺杂的碳材料。载体的纳米结构和极性表面有利于通过吸附和限域作用抑制PSs的扩散,且碳材料的优异导电性有利于其在正极和隔膜中的应用。Yang等[29]首次报道了SACs在锂硫电池中的应用。在该研究工作中,作者提出了Fe-N-C SAC可加快硫及其放电产物之间的可逆转化。近年来,多种SACs被应用于锂硫电池的正极[30-31]、隔膜[32-33]、电解液[34]中,以提高硫物种之间的氧化还原反应动力学,进而提高锂硫电池的电化学性能。
本文对近年来SACs在锂硫电池中的应用进行了综述。以影响催化剂性能的几个关键因素为主线,对该领域的研究成果进行了分类总结和讨论,并对SACs在锂硫电池领域的未来进行了展望。
1 SACs的活性中心金属原子
金属原子作为SACs的活性中心,对硫物种之间的转化反应动力学影响最大。Yang等[29]首次将SAC引入到锂硫电池体系。他们将Fe单原子负载到多孔氮掺杂的碳材料中,并将所得的Fe-N-C SAC(FeSA) 用作正极硫载体,实现了锂硫电池的高倍率性能和循环稳定性。这一方面归因于FeSA与PSs之间的强亲和力以及PSs在介孔碳微球上的物理吸附,从而限制了PSs的移动和穿梭;另一方面,FeSA可以作为电催化剂,加速可溶性PSs与不溶性的Li2S纳米粒子之间的氧化还原反应过程的动力学。如表1所示,目前,锂硫电池领域内报道过的SACs,活性中心均为第四周期的过渡金属,即Fe、Ni、Co、Mn、V。这主要是因为这些金属原子的d轨道未满,有利于通过成键作用活化底物。另一方面,这几种金属的价格便宜,对环境友好。从报道过的有限的几种金属活性中心也可以看出,SACs在锂硫电池领域的研究还有很大的潜力。
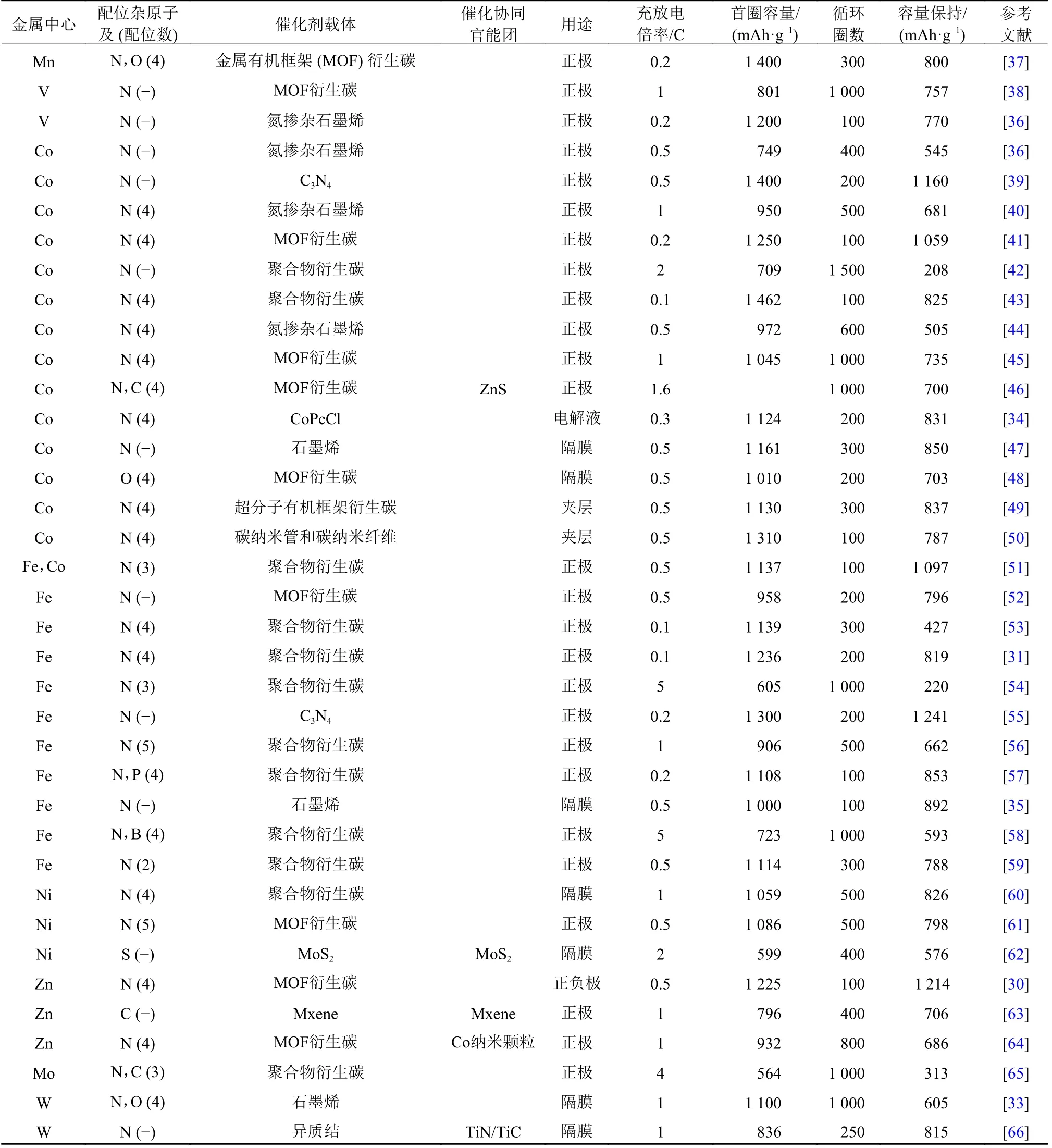
表1 报道过的SACs在锂硫电池中的应用情况总结Table 1 A summary of SACs for Li-S battery application
少数研究者对比了具有不同金属原子的SACs的催化性能。Zhang等[35]制备了氮掺杂石墨烯负载的Fe、Co、Ni SACs (FeSA、CoSA、NiSA) ,并将其应用于锂硫电池的隔膜涂层中。电化学测试结果发现,Fe-SA催化剂对应的电池展示出更低的充放电电压差和更好的循环稳定性。PSs的吸附实验结果显示,Fe-SA、CoSA、NiSA催化剂对Li2S6的吸附量分别为4.10、3.05、2.97 μmol·m-2,即FeSA吸附量最高 (见图1 (a) ) 。恒电流充放电曲线在比容量600 mAh·g-1时的充放电电压对比显示,FeSA电压间隙最小 (见图1 (b) ~ (c) ) ,说明FeSA相比其他SACs具有更佳的电化学活性。原位拉曼测试结果表明,使用单纯PP隔膜的电池在放电结束后仍然能观察到PSs的峰,而在具有FeSA修饰隔膜的电池中未检测到 (见图1 (d) ~ (e) ) ,表明其对PSs的氧化还原反应动力学具有促进作用。
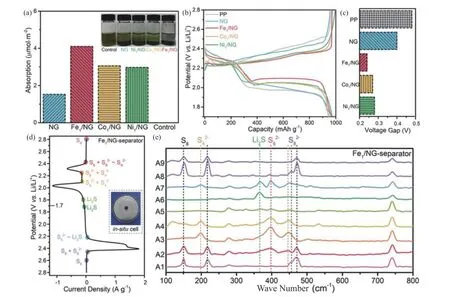
图1 (a) 不同材料对Li2S6吸附的电化学滴定结果(附图:12 h后Li2S6溶液的数码照片);(b) 0.5 C下的Li-S电池充放电曲线;(c) 600 mAh·g-1时不同隔膜的锂硫电池电压间隙;(d) 使用PP隔膜或FeSA隔膜的锂电池循环伏安(CV)曲线(附图:原位拉曼电池的数码照片); (e) 在(d) 中不同电压下,Fe/ NG修饰的锂硫电池的原位拉曼光谱[35]Fig.1 (a) Electrochemical titrations of the Li2S6 adsorption on different material (Inset: digital photo of the Li2S6 solution after 12 h) ;(b) Charge-discharge curves of the Li-S batteries at 0.5 C; (c) Voltage gaps of the Li-S batteries with various separators at 600 mAh·g–1; (d) Cyclic voltammetry (CV) profiles of the Li-S cell with the PP separator or the FeSA-modified separator (Inset:digital photo of the in situ Raman cell); (e) In situ Raman spectra of the Li-S cell with the Fe/NG-modified separator at different voltages as indicated in (d) [35]
SACs对硫物种之间的转化反应动力学的影响不仅体现在对PSs的催化转化上,还体现在对Li2S的催化转化中。Cui等[36]通过理论计算研究了一系列SACs对Li2S沉积能垒的影响。结果表明,单原子V负载到氮掺杂石墨烯上制得的催化剂 (SAV@NG) 对应的Li2S沉积能垒最低。实验结果表明,将SAV@NG用于锂硫电池正极,实现了高硫含量 (质量分数80%) 、快速反应动力学 (在3 C倍率下,容量为645 mAh·g-1)和长循环稳定性 (在0.5 C下容量衰减率为0.073%)。优异的电池性能归因于单原子V有效地捕获溶解PSs,并提高了硫物种之间的转化反应动力学,尤其是固态Li2S在放电和充电过程中的形成和沉积。
2 SACs的局部配位环境
SACs的金属单原子并不是简单地物理分散于载体表面,而是与载体表面的杂原子通过化学键键合配位的。目前,大多数SACs中的金属和载体中的杂原子是通过经典的四配位体系构建的。然而,近年来的研究表明,经典四配位体系中对称结构导致的对称电子分布不利于某些反应的进行[67-68]。诸多电催化反应证明,通过调整杂原子的配位数和配位杂原子的种类可以改变金属原子的电子状态,进而影响SACs的电催化性能[69]。
2.1 配位数的影响
Shang等[70]通过密度泛函理论 (Density Functional Theory, DFT) 对Fe、N-共掺杂石墨烯 (FeNx,x=1,2,3,4) 的锚定机理以及Li2Sn在FeNx上的吸附进行了研究。计算结果表明,由于Fe和S原子发生强烈的轨道杂化并形成化学键,FeNx对Li2Sn具有强亲和力。这将有利于抑制Li2Sn(n=4,6,8) 的溶解和扩散,以及Li2S和Li2S2的均匀成核。此外,计算结果表明,随着N原子配位数的增加,Fe-N和Fe-C键长变短,有利于增强Fe-N和Fe-C键的键能,进而提高Fe原子在掺杂体系中的稳定性。
Chen和Wang等[56]对比了Fe-N4和Fe-N5配位结构的Fe-N-C SACs材料在锂硫电池正极的应用。可视化PSs吸附和溶液紫外可见光谱 (Ultraviolet and visible spectrophotometry, UV-vis) 测试结果表明,具有Fe-N5配位结构的SAC对Li2S6的吸附性能优于Fe-N4配位结构的SAC (见图2 (a) )。电催化性能测试结果表明,Fe-N5-C材料表现出明显优于Fe-N4-C材料的电流响应和氧化还原电势差 (见图2 (b) );过饱和的Fe-N5-C活性位点可以更有效地降低PSs转化和Li2S成核的能垒 (见图2 (c) ),从而加速电化学反应的动力学。另外, Li2S到PSs的氧化过程中,Fe-N5-C的线性扫描伏安法 (Linear Sweep Voltammetry, LSV) 曲线显示出-0.49 V的最低起始电位和最高的电流响应 (见图2 (d) ),表明其对固体Li2S到PSs的反应动力学具有更优的催化作用。

图2 (a) N-C、Fe-N4-C、Fe-N5-C材料吸附Li2S6实验的溶液紫外可见光谱 (附图:Li2S6吸附试验的数码照片) ;(b) 不同电极的Li2S6对称电池的CV曲线;(c) N-C、Fe-N4-C和Fe-N5-C的Li2S沉积曲线;(d) N-C、Fe-N4-C和Fe-N5-C的Li2S氧化LSV曲线[56]Fig.2 (a) UV-vis spectra of Li2S6 solution soaked by N-C, Fe-N4-C, and Fe-N5-C (Inset: digital photographs of Li2S6 adsorption test) ;(b) CV curves of Li2S6 symmetric cells with different electrodes; (c) Li2S precipitation profiles of N-C, Fe-N4-C, and Fe-N5-C;(d) LSV curves of Li2S oxidization of N-C, Fe-N4-C, and Fe-N5-C[56]
2.2 配位原子种类的影响
目前,绝大多数的SACs是通过载体上的N原子与金属原子配位的。通过改变金属配位原子,例如,用B,C,O,S,P原子替换N,可调节金属原子的电荷分布,从而改变活性金属中心的电化学性质,进而影响催化性能。
Sun等[57]构建了多孔氮掺杂碳负载的FeSANx@PNC和多孔氮磷共掺杂碳负载的FeSAPN@PNC,其中FeSA-PN@PNC的配位结构为FeN3P1。DFT计算结果表明, FeN3P1的d波段中心高于FeN4。较高的d波段中心有利于提高反键轨道能量,从而导致更强的PSs亲和性和更高的催化活性。FeSA-Nx@PNC表面对Li2S和Li2S6的结合能分别为-2.19 eV和-1.16 eV,而FeSA-PN@PNC表面的结合能则急剧增加到-2.58 eV和-1.49 eV,表明FeSAPN@PNC对Li2Sx的锚定能力更强。另外,基于FeSAPN@PNC和FeSA-Nx@PNC计算得到的Li2S分解的能垒分别为1.52 eV和1.64 eV,说明FeSA-PN@PNC使Li2S分解能垒降低,从而有利于硫氧化反应的动力学。以上结果表明,用1个P原子取代FeN4配位体系中的一个N原子有利于增强对硫物种的吸附,并促进硫物种之间氧化还原反应的动力学。
Zhang等[65]构建了Mo-N3、Mo-N2C1和 Mo-N1C2三种SACs。Mo-N3结构d波段中心离费米能级最近,Mo-N1C2结构中d波段中心离费米能级最远 (见图3 (a) ~ (c) ) 。作者认为,Mo-N3结构对PSs的强吸附作用可能导致被吸附的PSs难以解吸,催化剂快速钝化,从而降低活性硫的利用率,而Mo-N1C2结构对PSs的吸附能力最弱,可能导致催化剂无法有效捕获PSs;Mo-N2C1结构可优化Mo与PSs之间的吸附和催化转化作用。吸附和电化学性能实验结果表明,Mo-N2C1实现了在电化学反应循环过程中对PSs的吸脱附以及转化。通过Mo K-edge X射线吸收光谱 (X-ray Adsorption Spectroscopy, XAS) 表征Mo-N2C1在充放电循环前后的演化过程,发现原始样品和充电样品的光谱形状有很好的重叠 (见图3 (d) ) ,表明Mo-N2C1具有很好的催化稳定性。
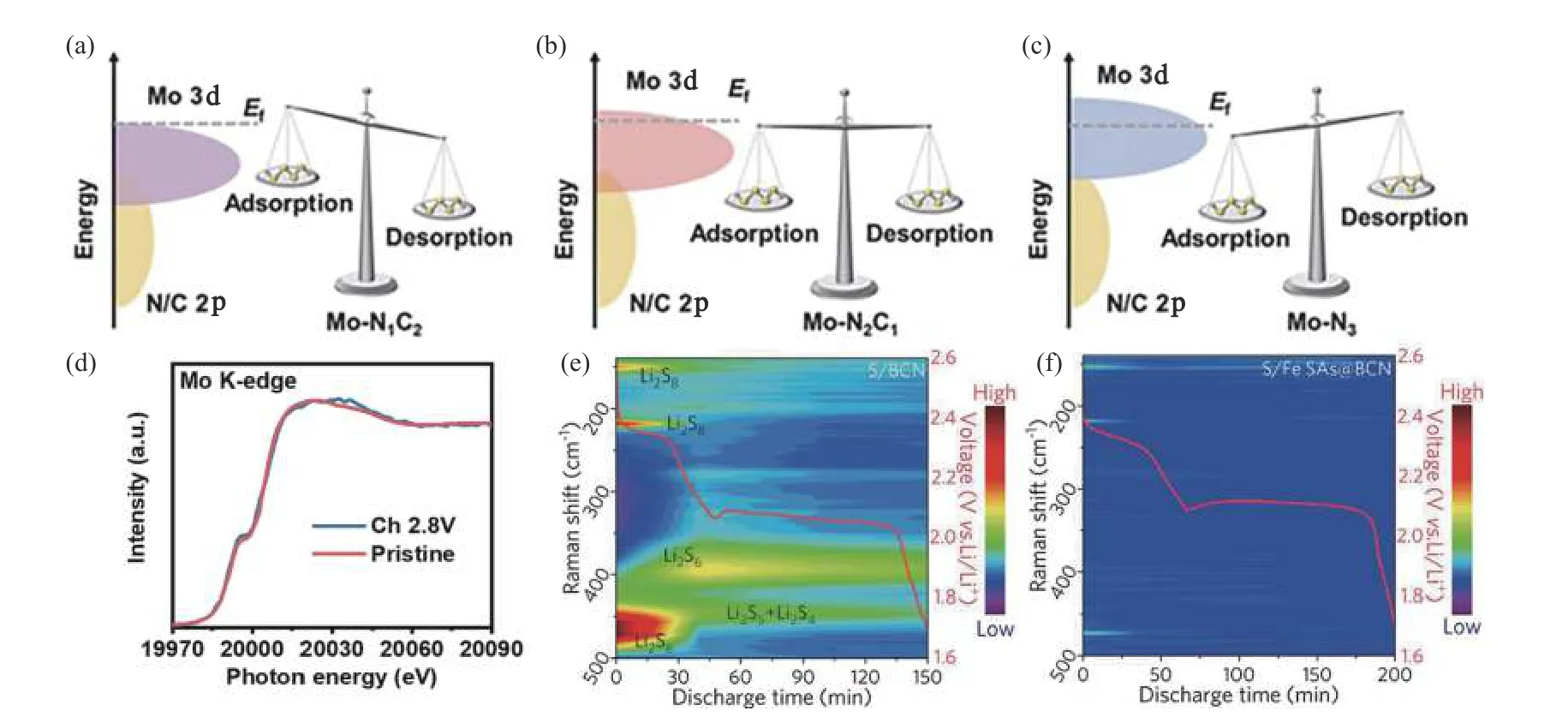
图3 (a)~(c) 不同结构的d波段中心位置与PSs吸附-解吸能力关系示意图;(d) 循环前后Mo-N2C1的K-edge XAS光谱[65];(e) 不同放电状态下S/BCN电极的原位时间分辨拉曼轮廓图;(f) 不同放电状态下的S/SAFes@BCN电极的原位时间分辨拉曼轮廓图[58]Fig.3 (a)~(c) Schematic illustration of the relationship between d-band center positions and PSs adsorption-desorption capacities for different structures; (d) Mo K-edge XAS spectra of Mo-N2C1 before and after cycling [65]; (e) In-situ time-resolved Raman contour plots S/BCN electrodes at different discharge states ; (f) In-situ time-resolved Raman contour plots S/SAFes@BCN electrodes at different discharge state[58]
Wu和Zhang等[58]构建了硼、氮共配位铁SAC(SAFe @BCN) 。在非对称电子结构N2-Fe-B2中,氮原子作为电子受体,硼原子作为电子供体,氮原子更容易从硼原子处得到电子,使金属中心铁原子保留3d电子。因此,N原子被B原子部分取代有利于金属原子周围的电子离域分布,从而增强对PSs的亲和力,加速PSs转化过程中的电荷转移。原位拉曼光谱实验结果显示,在放电过程中使用BCN材料的电池可明显检测到PSs产物,而使用SAFe @BCN材料的电池中无明显的PSs信号 (见图3 (e) ~ (f) ),证实了该SAC对硫物种的强亲和力及其对硫还原过程的高电催化活性。
3 催化剂载体的影响
3.1 碳材料作为SACs的载体
SACs与载体中的杂原子通过键合分布于载体表面。因此,催化剂载体的属性(如杂原子浓度及种类)会对金属的锚定和活性中心的电子特性产生影响。另一方面,在锂硫电池的应用中,催化剂载体难免会与PSs接触,载体的属性(如与PSs的亲和性)亦会对“穿梭效应”产生影响。锂硫电池中硫和硫化锂固有的导电性差的问题导致了电池反应动力学缓慢、硫利用率低等问题。碳材料优异的导电性使其成为SACs载体的理想材料。丰富多样的碳材料为SACs载体材料的选择提供了广阔的空间。
根据碳材料的结构形貌可将其划分为零维(0D) 、一维 (1D) 、二维 (2D) 、和三维 (3D) 材料。目前,研究者们报道过多种形貌的碳材料作为锂硫电池用SACs的载体。Liu等[52]以经典的MOF材料ZIF-8为前驱体,制备了负载FeSA的氮碳纳米笼材料,并将其用作硫正极载体。纳米笼丰富的多孔结构和极性位点有效抑制了PSs的穿梭效应,同时分散均匀的FeSA有效促进了电化学反应过程中硫物种之间的转化。Chen等[71]以ZIF-7为前驱体制备了负载ZnSA的高度有序氮掺杂碳纳米管阵列作为锂硫电池的正极硫载体。规则的1D碳纳米管结构促进了在高硫负载下的电池反应的快速传质,而碳纳米管上负载的丰富的单原子活性位点有效加速了PSs的转化。Ji 和Wu等[40]将CoSA负载于氮掺杂石墨烯上,并将其用作正极硫载体。该SAC材料实现了电池在高硫负载(6.0 mg·cm-2) 下的循环稳定性,在100次循环中每个循环的容量衰减率为0.029%。这得益于单分散钴原子可以触发PSs的表面介导反应,而Co-N-C配位中心作为双功能电催化剂,可分别在放电和充电过程中促进Li2S的形成和分解。
Sun等[59]使用硅藻土为模板制备了负载FeSA的3D结构氮碳材料 (3DFeSA-CN) ,并将其用于锂硫电池正极。作者对比了3DFeSA-CN与非3D结构的负载FeSA的氮碳材料 (FeSA-CN) 对硫氧化还原反应的电催化作用。在Li2S的沉积成核实验中,基于3DFeSA-CN的电池更快达到电流峰值,意味着Li2S的沉积和溶解更为快速。在对Li2S6的催化转化实验中,与FeSA-CN材料相比,3DFeSA-CN材料的电池显示出更尖锐的氧化还原峰和更小的电压极化,表明其对硫氧化还原反应有更强的催化加速作用。对比相应峰的Tafel曲线,S@3DFeSA-CN电极具有更高的斜率,表明其更有利于锂离子扩散以及硫物种之间的氧化还原反应。此外,3DFeSA-CN材料继承了硅藻土模板独特的分层结构,具有丰富的多级孔通道。这种多级多孔结构不仅可以实现高硫负载,还可以缓解电化学循环时的体积变化。对循环后的电极进行扫描电子显微镜 (Scanning Electron Microscope , SEM)表征,结果显示,S@FeSA-CN阴极表面会产生大量硫化物和裂纹,而S@3DFeSA-CN表面相当光滑,没有明显的开裂和团聚 (见图4) 。

图4 0.2 C下循环200次后电极表面的扫描电镜图像:(a)~(b) S@3DFeSA-CN;(c)~(d) S@FeSA-CN;(e)~(f) S@CN[59]Fig.4 SEM images of the electrode surface after 200 cycles at 0.2 C: (a)~(b) S@3DFeSA-CN;(c)~(d) S@FeSA-CN; (e)~(f) S@CN[59]
3.2 极性材料作为SACs的载体
碳材料的非极性特性导致材料与极性PSs的相互作用较弱,限制了其在循环过程中束缚和限制PSs的能力[72]。因此,少数研究者使用了极性材料作为SACs的载体。
MXene材料,即2D过渡金属碳化物/氮化物,由于优异的电子导电性、大比表面积和结构可调性等特点在储能领域备受关注[73]。2015年,Nazar等[74]率先将MXene材料应用于锂硫电池阴极。此后,多种MXenes材料开始应用在锂硫电池领域[75-78]。Yang等[63]将单原子Zn引入到MXene材料Ti3AlC2中,制备了SAZn-MXene层状材料。通过高角度环形暗场扫描透射电子显微镜 (High-Angle Annular Dark-Field Scanning Transmission Electron Microscopy, HAADFSTEM) 图像 (见图5 (a) ) 揭示了MXene纳米片上高度分散的亮点 (Zn原子) 。在PSs吸附实验中,SA-Zn-MXene呈现出优异的吸附性能 (见图5 (b) ) ,这得益于SA-Zn-MXene与PSs之间的高结合能。CV曲线的对比显示,与MXene相比,SA-Zn-MXene可以实现更高的电流密度 (见图5 (c) ) ,表明SA-Zn-MXene可以更有效地加速PSs的转化反应动力学。进一步的DFT计算表明,SA-Zn-MXene可大幅度降低限速步骤的能量势垒 (Li2S2到Li2S) (见图5 (d) ) 。因此,SAZn-MXene不仅可以有效吸附PSs,还可以促进硫物种之间转化反应的动力学,从而实现锂硫电池出色的电化学性能。

图5 (a) 单原子锌引入MXene的制备简图(右图是SA-Zn-MXene经像差校正后的HAADF-STEM图像);(b) SA-Zn-MXene、MXene和super P的紫外/可见光谱和视觉吸附测试;(c) SA-Zn-MXene、MXene和super P的Li2S6对称电池的CV曲线;(d) 多硫化锂在SA-Zn-MXene和MXene上的吉布斯自由能谱[63]Fig.5 (a) The illustration of the fabrication of single atom zinc implanted MXene (SA-Zn-MXene) (the right figures in (a) are the aberration-corrected HAADF-STEM images of SA-Zn-MXene); (b) Visual adsorption tests of SA-Zn-MXene, MXene, and super P with their corresponding UV/vis spectra; (c) CVs of Li2S6 symmetric cells of SA-Zn-MXene, MXene, and Al foil; (d) The Gibbs free energy profiles of lithium polysulfides on SA-Zn-MXene and MXene[63]
金属硫化物由于具有优良的机械性能、热稳定性以及丰富的边缘活性位点,在电催化领域得到了广泛关注[79]。Xu和Mai等[62]将单原子Ni引入到层状MoS2材料中,并将制得的SAC (SANi-MoS2) 用于锂硫电池的隔膜修饰。多种表征结果证明Ni单原子被成功引入并均匀分布在MoS2结构中 (见图6 (a) ~ (c) ) 。CV测试结果表明,相比MoS2@PP和PP隔膜组装的电池,SANi-MoS2@PP隔膜组装的电池的还原电位更接近PSs标准还原电位,验证了SANi-MoS2材料对PSs还原反应的促进作用。对于Li2S2/Li2S和PSs之间转变的氧化还原反应过程,SANi-MoS2@PP隔膜的Tafel斜率比其他两种隔膜的Tafel斜率都低。此外,在Li2S6对称电池的催化性能测试中,SANi-MoS2@PP隔膜组装的电池响应电流最大。以上结果均证明SANi-MoS2材料有助于提高硫物种之间的转化反应动力学。使用该材料修饰的锂硫电池隔膜实现了优异的电化学性能:在2 C的测试倍率下400次循环的每圈容量衰减率为0.01%;硫负载为7.5 mg·cm-2,电池在0.2 C下循环50次后仍保持5.9 mAh·cm-2的容量。

图6 (a) Ni-MoS2纳米片的HAADF-STEM图像及其对应的EDS图谱;(b) Ni-MoS2纳米片的原子分辨率图像;(c) Ni-MoS2纳米片的傅里叶变换后的原子分辨率图像[62]Fig.6 (a) HAADF-STEM image of Ni-MoS2 nanosheets, and its corresponding EDS mapping; (b) Atomic resolution picture of the Ni-MoS2 nanosheets and (c) related FFT-filtered atomic resolution image[62]
4 多催化活性中心的协同
受当前SACs合成技术的限制,载体材料上金属原子的比例非常低[80](通常质量分数小于10%) ,这无疑会影响催化剂的活性。为了提高活性中心的数目,研究者们尝试通过多催化活性中心协同作用来提高SACs的催化性能。
Pan和Wu等[64]在氮掺杂多孔碳纳米片接枝的碳纳米管上同时引入了Co纳米颗粒和单原子Zn (Co/SAZn@N-C/CNTs) ,并与单独引入Co纳米颗粒 (Co@NC/CNTs) 和单独引入单原子Zn的材料 (SA-Zn@N-C)做了催化活性对比。通过组装Li2S6对称电池进行CV测试以评价材料对PSs的催化效果。Co/SAZn@N-C/CNTs电极在0.404 V (A峰: Li2S6的还原) 、0.031 V (B峰: Li2S/ Li2S2氧化) 、0.309 V (C峰: Li2S6氧化为S) 和0.022 V (D峰: S还原为Li2S6) 处显示出4个明显的氧化还原峰 (见图7 (a) ~ (d) ) ;Co@N-C/CNTs和SA-Zn@N-C可以观察到4个峰,但峰的电流密度较低;氮掺杂碳材料 (N-C) 只显示2个峰。这说明在Co/SA-Zn@N-C/CNTs复合材料中原子分散的Zn-N4和Co纳米颗粒的协同作用可以显著加速PSs的电化学转化。恒电位Li2S成核测试结果表明,Co/SAZn@N-C/CNTs使Li2S沉淀的累积容量高达302.9 mAh·g-1(见图7 (e) ~ (h) ) ,远远大于Co@N-C/CNTs(263.2 mAh·g-1) 、SA-Zn@N-C (223.7 mAh·g-1) 和N-C(156.2 mAh·g-1) ,进一步证明Co/SA-Zn@NC/CNTs对Li2S沉淀的催化活性最好。DFT计算结果表明,Co/SA-Zn@N-C/ CNTs中钴纳米粒子和Zn-N4的耦合会诱导出最佳的电子结构,能有效提高催化转化反应的能力,尤其是Li2S的形成和分解的能力。

图7 (a) Co/SA-Zn@N-C/CNTs、(b) Co@N-C/CNTs、(c) S@SA-Zn@N-C和(d) S@N-C在-1 V~ 1 V扫描速率为0.1 mV·s-1时对称电池的CV曲线,Li2S8作为阴极电解质;(e) Co/SA-Zn@N-C/CNTs、(f) Co@N-C/CNTs、(g) SA-Zn@N-C和(h) N-C在2.05 V下的恒电位放电曲线[64]Fig.7 CV curves of symmetric cells for (a) Co/SA-Zn@N-C/CNTs, (b) Co@N-C/CNTs, (c) S@SA-Zn@N-C and (d) S@N-C from -1 to 1 V at a scan rate of 0.1 mV·s-1.Potentiostatic discharge profiles at 2.05 V with Li2S8 catholyte of (e) Co/SA-Zn@N-C/CNTs,(f)Co@N-C/CNTs, (g) SA-Zn@N-C and (h) N-C[64]
Xu和Mai等[51]将Fe-Co双金属引入碳纳米球构造了负载Fe-Co双金属的SAC (Fe-CoNC) 。Li2S成核试验中,Fe-CoNC复合电极电池的响应电流峰值时间(1 891 s) 早于其他复合电极电池的响应电流峰值(FeNC (2 626 s) ,CoNC (2 838 s) ,NC (3 158 s) ),且成核容量更高 (Fe-CoNC为409 mAh·g-1,高于FeNC(195 mAh·g-1)、CoNC (190 mAh·g-1) 和NC (183 mAh·g-1) )。这表明Fe-CoNC可以更好地改善Li2S成核动力学,促进Li-S电池的液固反应。另一方面,在Li2S溶解实验中,Fe-CoNC的溶解容量 (1 246 mAh·g-1) 高于CoNC (1 174 mAh·g-1)、FeNC (780 mAh·g-1) 和NC(674 mAh·g-1),表明Fe-CoNC对Li2S去核化具有更高的催化活性。此外,单独对比Co-NC与Fe-NC材料的成核容量、响应电流以及溶解容量,结果表明FeNC更有利于Li2S成核,而CoNC更倾向于催化Li2S的溶解。以上结果证明了Fe-Co双单原子的协同效应,即Fe单原子更有利于Li2S成核,而Co单原子加速Li2S分解,从而改善Li-S电池的双向氧化还原反应动力学。
Zhao等[46]将极性ZnS纳米颗粒和Co-N-C结构的SAC嵌入到高度定向的大孔导电框架中构建了双端结合 (Double-end Binding, DEB) 位点的催化剂 (3domsh/ZnS/Co-N-C) 并将其用作正极硫载体。DFT计算结果表明,由于Co-N-C SAC和极性ZnS的协同作用,该DEB材料与PSs的结合强度都高于单端结合(Single-end Binding, SEB) 位点材料,这有利于抑制PSs的穿梭。原位高能X射线衍射 (High Energy X-ray Diffraction, HEXRD) 表征显示 (见图8 (a) ) ,DEB位点能促进固体Li2S2和液体PSs之间的快速有效转化。对以1 C的倍率循环100次后的电池S阴极进行S-二次离子的飞行时间二次离子质谱 (Time-of-flight Secondary Ion Mass Spectrometry, ToF-SIMS) 测试,结果表明(见图8 (b)~(c) ),循环后的科琴黑 (Ketjen Black,KB) /S和3d-omsh/纯碳/S阴极表现出的PSs穿梭引起的S迁移层强度最高,而3d-omsh/ZnS/S材料和3domsh/Co-N-C/S材料的S迁移层强度峰值变弱,3domsh/ZnS/Co-N-C/S材料没有观察S迁移层,证实了DEB位点在固定PSs方面的有效性。对循环后的锂金属进行ToF-SIMS分析,结果显示(见图8 (d) ) ,在使用KB和未负载ZnS和SACo的三维材料做正极的电池中鉴定出S迁移层,表明有严重的穿梭效应和锂金属腐蚀;使用SEB位点的材料时,循环后锂金属阳极表面S元素的强度变弱但仍然可见,而在使用DEB位点的材料时,只有非常微弱的S信号可以在循环后锂金属阳极的表面上找到,证明DEB位点配置策略在抑制PSs穿梭上的有效性。

图8 (a) 3d-omsh/ZnS, Co-N-C /S阴极在0.1 C下的充放电曲线和相应的原位HEXRD图;(b) 在1 C循环100次后S-二次离子的ToFSIMS深度分布图;(c) 在1.0 C循环100次后的S-二次离子的ToF-SIMS深度分布的3D渲染图: (i) KB/S,(ii) 3D -omsh/纯碳/S,(iii) 3D-omsh/ZnS/S,(iv) 3D-omsh/ Co-N-C /S和 (v) 3D-omsh/ZnS, Co-N-C /S阴极;(d) 在循环的Li金属阳极表面的ToF-SIMS S-元素映射图: (i) Li-KB/S,(ii) Li-3D-omsh/纯碳/S,(iii) Li-3D-omsh/ZnS/S,(iv) Li-3d-omsh/Co-N-C/S和 (v) 在1 C下100个周期后的Li-3d-omsh/ZnS, Co-N-C/S电池[46]Fig.8 (a) Charge/discharge curve of the 3d-omsh/ZnS, Co-N-C/S cathode at 0.1 C and the corresponding in situ HEXRD patterns; (b) ~(c)ToF-SIMS depth profiles and 3D rendering of S-secondary ion distribution of cycled: (i) KB/S, (ii) 3d-omsh/pure carbon/S, (iii) 3domsh/ZnS/S, (iv) 3d-omsh/Co-N-C/S and (v) 3d-omsh/ZnS, Co-N-C/S cathodes after 100 cycles at 1 C; (d) ToF-SIMS S-element mappings on the surface of cycled Li metal anode of: (i) Li-KB/S, (ii) Li-3d-omsh/pure-carbon/S, (iii) Li-3d-omsh/ZnS/S, (iv) Li-3domsh/Co-N-C/S and (v) Li-3d-omsh/ZnS, Co-N-C/S cells after 100 cycles at 1 C[46]
5 总结与展望
本文对近年来SACs在锂硫电池中的应用进行了综述。以影响催化剂性能的几个主要关键参数为主线,分别从催化剂的金属活性中心、配位环境、载体属性、催化协同几个方面分类讨论总结了近年来的研究进展。总的来说,目前,SACs在锂硫电池中的应用研究工作还比较有限,该研究方向还有广阔的研究空间和潜力。结合已有的研究结果,作者认为,未来锂硫电池用SACs的研究可侧重在以下几个方面:
(1) 催化活性中心。SACs的电催化性能与金属原子的种类及其配位环境密切相关。目前涉及到的金属原子种类很少,主要是第四周期的过渡金属,如Fe、Ni、Co、Mn、V。未来在金属原子的选择上还有很多空间。另一方面,除了利用当前研究最多的N原子来配位键合金属原子外,其他杂原子如B、 O、 P、 S等可尝试作为金属配位的杂原子,调节金属的配位环境,从而调节催化性能。
(2) SACs的载体属性(如形貌、比表面积、导电性、溶解性等) 对催化剂的性能具有显著影响。考虑到硫及其放电产物的绝缘性,碳材料目前是构建SACs的首选载体。少量的极性载体,如MXenes和金属硫化物,也被证明是出色的SACs载体材料。结合实际需求,选择和设计制备具有特定属性的SACs载体材料是很有必要和意义的。
(3) 受限于单原子催化剂的制备技术,SACs中的金属载量很低,这将大大限制催化剂的性能。如何在保持原子级分散的前提下提高金属载量依然是未来努力的重点。另外,引入已有的其他类型的催化剂(如异质结、量子点等) 与SACs协同作用也是提升催化性能的方案之一。
(4) 硫物种之间的氧化还原反应是多种中间产物的双向转化。目前,SACs与硫物种之间的催化作用机理仍然不清晰。结合先进的原位表征技术监测SACs与硫物种之间的相互作用,有助于揭示SACs及其催化性能之间的构效关系,探索详细机理,对进一步有针对性地构建SACs材料具有重要意义。
(5) 目前SACs的研究大部分处在基础研究阶段,如在半电池中研究其对锂硫电池的容量和寿命的提升作用。顺应商业化要求,要研发低成本、高硫负载量、贫电解液条件下的高性能锂硫全电池,还有很长的路要走。
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 06:42:32
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09 06:12:08
装备制造技术(2020年4期)2020-12-25 05:25:52
当代陕西(2019年6期)2019-04-17 05:04:10
电源技术(2015年2期)2015-08-22 11:28:30
电源技术(2015年5期)2015-08-22 11:17:54
无机化学学报(2014年4期)2014-02-28 17:31:08
电源技术(2014年9期)2014-02-27 09:03:46
