
巨噬细胞极化与动脉粥样硬化斑块稳定性的研究进展①
2023-11-13 09:29谢燕飞江西中医药大学医学转化中心南昌330004
中国免疫学杂志 2023年10期
刘 理 谢燕飞 余 军 (江西中医药大学医学转化中心,南昌 330004)
动脉粥样硬化是一种慢性炎症性疾病,其主要病变表现为部分动脉脂质沉积,并伴有平滑肌细胞和纤维基质增生,逐渐发展为动脉粥样硬化斑块,斑块破裂往往伴随纤维帽变薄、脂质池增大、炎症活动增加、蛋白水解酶含量增加等现象[1]。巨噬细胞可分为M1 型和M2 型,分别释放促炎因子及抑炎因子,促炎因子可促进斑块形成,反之抑炎因子减缓斑块形成。同时,巨噬细胞两种表型可在一定条件下相互转换[2]。研究表明,斑块病变处存在大量不同表型巨噬细胞,稳定斑块中以M2 型巨噬细胞为主,不稳定斑块中以M1 型巨噬细胞为主[3]。综上,巨噬细胞及其极化状态对斑块的稳定性具有重大影响,以极化为切入点可能是治疗动脉粥样硬化的有效途径。因此,本文从巨噬细胞表型、巨噬细胞极化对斑块的影响以及表型机制调节等方面进行综述。
1 巨噬细胞极化
巨噬细胞极化主要受微观环境影响,不同刺激可导致巨噬细胞向不同表型转化。目前主要将巨噬细胞分为两大类,即促炎M1 型及抗炎M2 型,前者由Th1 细胞因子诱导,后者由Th2 细胞因子诱导[4]。两种细胞亚型功能、表面标志物、趋化因子等方面各不相同(表1)。

表1 巨噬细胞不同表型及其特点Tab.1 Phenotypes and characteristics of macrophage
M1 型巨噬细胞主要发挥促炎作用,能产生TNF-α、IL-6 等炎症因子及CXCL9、CXCL10 等标志物[5]。M1 型巨噬细胞还能产生活性氧、一氧化氮合酶等物质,诱导组织损伤并阻碍伤口愈合[6]。
M2 型巨噬细胞可进一步分化为M2a、M2b、M2c、M2d 4 种亚型。M2a 巨噬细胞由IL-4、IL-13 诱导,具有组织重塑、内吞功能;M2b 巨噬细胞由免疫复合物与IL-1β 或脂多糖联合诱导,主要发挥免疫调节作用;M2c 巨噬细胞由IL-10、TGF-β 或糖皮质激素诱导,发挥细胞外吞作用;M2d巨噬细胞由TLR以及腺苷A2A受体激动剂共同诱导[5,12-13]。所有M2型巨噬细胞均能发挥一定抗炎效应,主要表现为IL-12 低表达、IL-10 和TGF-β 高表达,但每种亚型都有其特定标志物,并表现不同特性(表1)。
除M1、M2型巨噬细胞外,近年又陆续发现了其他不同亚型巨噬细胞,如M(Hb)、Mhem、Mox、M4等,其中Mhem 是对HA-mac 重新定义的结果,具有抑制氧化应激、抗炎效果(表1)[14-21]。
2 不同表型巨噬细胞在斑块中的定位
动脉粥样硬化往往伴随炎症反应,炎症因子可刺激巨噬细胞分泌MMP-2和MMP-9,降解细胞外基质,影响纤维帽厚度,从而影响斑块稳定[22]。此外,研究发现,ApoE-/-MMP10-/-小鼠斑块中M1 型巨噬细胞极化标志物IL12a、NOS2和TNF-α表达减少,小鼠炎症程度较低,斑块表型更稳定[23]。随着对斑块的深入研究发现,斑块中存在不同巨噬细胞表型,且存在分布差异。斑块中M1、M2 型巨噬细胞数量均较外周正常组织明显增多。CHO等[3]在此基础上进一步研究斑块中巨噬细胞分型,发现稳定斑块中M2 型呈高分布,而不稳定斑块中以M1 型居多。与M2型相比,M1型多分布于内膜易破裂的肩部区,M1型巨噬细胞增多促进斑块破裂,导致血栓形成。
3 不同巨噬细胞表型对斑块的影响
3.1 M1 和M2 巨噬细胞 剪切力、维生素D 受体、LPS、IFN-γ 等均可诱导巨噬细胞向M1 型极化,并释放促炎因子如IFN-γ、IL-1β、TNF-α。炎症因子可破坏内皮细胞,促进平滑肌细胞、巨噬细胞凋亡,凋亡细胞堆积形成脂核,进一步加速斑块形成[7-8]。另外,研究证实M1型巨噬细胞释放的促炎因子TNF-α在动脉斑块中高表达,尤其在不稳定斑块内膜与中膜[24]。研究发现,动脉粥样硬化病变中的M1 型巨噬细胞通过活化NF-κB 信号通路增加C-反应蛋白(C-reactive protein,CRP)表达,且斑块中M1 型巨噬细胞数与CRP 水平呈正相关[15]。因此,诱导巨噬细胞从M1 型向M2 型转变或许可减少CRP 的有害作用,并减缓粥样硬化斑块中的炎症反应。与M1 型巨噬细胞相反,M2 型巨噬细胞分泌的IL-10 等抑炎因子能增强斑块稳定性[25]。LIU 等[26]取颈动脉斑块患者和正常人外周血单核细胞(peripheral blood mononuclear cells,PBMCs)分化为巨噬细胞,检测两组PBMCs 源巨噬细胞基因表达差异,结果表明,与正常组比较,颈动脉斑块患者低表达LXRα,进一步过表达LXRα 发现,M2 型标志物CD163、CD206、Arg1表达升高,M1型标志物TNF-α、IL-6、IL-10表达降低。提示LXRɑ过表达促使PBMCs源巨噬细胞向M2型极化。
3.2 M4、Mox、Mhem、M(Hb)型巨噬细胞 研究者发现了与斑块稳定相关的M4 型巨噬细胞,这种表型的巨噬细胞由血小板趋化因子CXCL4 诱导[20]。OKSALA 等[21]进一步研究发现,人颈动脉斑块中所有M4 巨噬细胞标志物均与MMP12和组蛋白去乙酰化酶9(HDAC9)相关。表明M4 巨噬细胞可能是HDAC9和MMP-12在晚期人类斑块中表达的来源。
KADL 等[18]在小鼠斑块中发现了Mox 型巨噬细胞,与传统的M1 或M2 表型相比,这种巨噬细胞由氧化磷脂诱导,显示出弱的吞噬能力和趋化特性,氧化还原调节的转录因子Nrf2 参与调控其形成过程。这种类型巨噬细胞主要通过上调Nrf2、增加Hmox1 蛋白表达及转录、膜铁转运蛋白(FPN)转录引起铁潴留,从而促进动脉粥样硬化发展[19]。血红素(hemoglobin,Hb)刺激的巨噬细胞也称为M(Hb)型巨噬细胞,这种巨噬细胞高表达MR/CD163[14]。FINN 等[14]发现,与泡沫细胞相比,M(Hb)型巨噬细胞内脂质沉积明显减少。同时,M(Hb)型巨噬细胞内含铁量减少,促使ROS减少,进一步使LXRα受体驱动胆固醇外排基因ABCA1表达增加,最终增加胆固醇外排,延缓动脉粥样硬化发展,与Mox型巨噬细胞铁表型促进铁潴留和脂质累积恰恰相反。另一种巨噬细胞——斑块出血区的Mhem 细胞可通过转录激活因子1(transcription factor 1,ATF-1)影响动脉粥样硬化发展。如BOYLE 等[15]指出斑块内出血区的HA-mac/Mhem共表达p-ATF-1、HO-1 以及LXR 的靶基因ApoE 和ABCA1,减少泡沫细胞变化,从而减轻斑块内出血。
综上,巨噬细胞极化与微环境紧密相关,极化状态以及极化后的分布对斑块稳定性尤为重要。因此,治疗动脉粥样硬化时可通过调节M1/M2 型巨噬细胞比例使巨噬细胞向M2 型极化,或诱导巨噬细胞向其他增加斑块稳定性的表型分化,延缓动脉粥样硬化性心血管疾病发生。此外,虽然M1 型及M2 型巨噬细胞对动脉粥样硬化的作用已明确,但M2 型巨噬细胞各亚型在动脉粥样硬化中的作用及具体机制还有待阐明。
4 巨噬细胞极化的调节机制
4.1 miRNAs miRNAs是一种小的、内源性的非编码RNA,长度为22~26 个核苷酸,广泛参与癌症、肥胖、动脉粥样硬化等多种疾病发生发展[27]。研究表明miR-33、miR-155、miR-223、miR-27 等均可影响巨噬细胞极化[28-31]。NAZARI-JAHANTIGH 等[31]研究发现,miR-155 在动脉粥样斑块及促炎型巨噬细胞中上调,进一步研究发现miR-155 通过抑制转录因子BCL-6 表达促进NF-κB 信号转导,增加趋化因子CCL2 表达,促进动脉粥样硬化发展。DONNERS等[32]将miR155-/-及野生型小鼠骨髓移植到LDLR-/-小鼠中,研究miR-155对动脉粥样硬化的作用,结果发现,与正常组相比,miR-155-/-组小鼠斑块面积增大,血脂水平升高,且动脉粥样病变处中性粒细胞、巨噬细胞增加,T 细胞减少,提示miR-155 具有抗动脉粥样硬化作用。OUIMET 等[30]为研究miR-33 与巨噬细胞炎症的关系,将骨髓来源巨噬细胞(BMDMs)分别用IFN-γ/LPS或IL-4处理24 h后检测miR-33水平,结果显示,与M2 型相比,M1 型巨噬细胞miR-33表达更高,提示miR-33 可能参与巨噬细胞M1 型极化,进一步过表达miR-33 后发现,M1 型巨噬细胞标志物(如IL-6、NOS2 和IL-1β)转录水平升高,同时M2 型巨噬细胞标志物CD206 和Fizz1/Retnla 转录水平降低。
miR-216a 可通过靶向Smad/NF-κB 信号通路发挥促炎作用[33]。YANG 等[34]在此基础上研究miR-216a对巨噬细胞极化的影响,发现miR-216a能够通过Smad/NF-κB 信号通路诱导巨噬细胞中端粒酶激活,并促进巨噬细胞向M1 型极化。由此看来,靶向miR-216a 开展治疗对稳定斑块、治疗动脉粥样硬化或将起到积极作用。
4.2 转录因子 STAT6作为STAT家族成员在细胞分化和细胞因子产生中发挥重要作用[35]。据报道,高脂喂食ApoE-/-小鼠建立动脉粥样硬化模型后,易损斑块中CD86、iNOS 表达升高,Arg-1、TGF-β 表达降低。进一步研究发现,易损斑块中STAT6 及磷酸化STAT6 表达降低,稳定斑块中STAT6 及M2 型标志物CD163上升[36]。STAT6在动脉粥样化中的作用在其他报道中也得到了证实。王凌等[37]研究脂连蛋白对动脉粥样硬化小鼠的作用,发现脂连蛋白可能通过上调p-STAT6 表达诱导脂肪组织巨噬细胞M2型极化,发挥抗动脉粥样硬化作用。
糖原合酶激酶-3α(glycogen synthase kinase-3α,GSK-3α)参与糖原合酶合成及细胞增殖、分化[38]。MCALPINE 等[39]建 立 了 骨 髓 特 异 性 敲 除GSK3α(LMαKO)的LDLR-/-小鼠模型,结果表明,LMαKO小鼠巨噬细胞中GSK3α 缺失可显著增强STAT3 和STAT6 磷酸化,最终减弱M1 型标志物CD36 表达,同时促进M2 型标志物Arg-1、Pgc1 表达,提示巨噬细胞GSK3α对斑块的稳定可能有影响。
转录因子Mafb 属于Maf 家族,Mafb 在造血骨髓细胞中高表达,对单核巨噬细胞谱系建立和维持起重要作用[40]。KIM 等[41]通过慢病毒技术将Mafb 导入小鼠骨髓来源巨噬细胞,检测不同表型巨噬细胞标志物表达,结果发现Mafb 抑制M1 型巨噬细胞标志物表达,促进M2 型标志物表达,进一步研究表明,Mafb 通过IL-4/STAT6 通路调节巨噬细胞向M2型极化,在RAW264.7 巨噬细胞中也得出了同样结果,提示Mafb 可能对动脉粥样硬化斑块进展起保护作用。其他转录因子如同源盒A5(homeobox A5,HOXA5)是同源盒转录因子之一,与巨噬细胞表型转换密切相关[41]。最近研究证实HOXA5 通过激活颈动脉转录辅激活子1(mediator subunit 1,MED1)促进动脉粥样硬化斑块中巨噬细胞向M2 型转换,延 缓 动 脉 粥 样 硬 化 进 展[42]。Krüppel 样 因 子4(KLF4)是一种锌指转录因子,属于KLF4 转录因子家族。已有研究表明KLF4 可调节巨噬细胞极化[43]。LIAO 等[44]研究卡利司汀(KS)这种组织激肽酶结合蛋白和丝氨酸蛋白酶抑制剂时,发现KS通过激活KLF4 调节巨噬细胞M1/M2 比例延缓ApoE-/-小鼠斑块形成速度。
4.3 核受体 过氧化物酶体增殖物激活受体γ(peroxisome proliferator-activated receptor-γ,PPAR-γ)是核激素受体家族中的配体激活受体,具有抗炎活性[45]。研究表明,PPAR-γ 能促进单核细胞向M2 型巨噬细胞分化[46]。此外,也有研究发现瑞舒他汀通过激活PPAR-γ 促进人外周血单核细胞中M2 型标志物CD206、IL-10、CCL18 表达增加,从而治疗动脉粥样硬化[47]。
NR4A 家族核受体Nurr1、Nur77 和NOR1 在人动脉粥样硬化病变的巨噬细胞中表达[48-49]。研究表明,LDLR-/-小鼠中Nur77缺失可促进巨噬细胞向M1型极化,从而导致动脉粥样硬化病变增加[49]。NOR1 高表达于人动脉粥样斑块中CD68+CD206+(M2 型巨噬细胞)富集区域,沉默NOR1 后,M2 型标志物CD206、IL-1Ra、CD200R、F13A1、IL-10 水平降低。另外,全基因组芯片分析显示,在M2 巨噬细胞中沉默NOR1后,MMP-9上调较为明显。提示NOR1可诱导人巨噬细胞向M2 型转化,并通过调控MMP-9影响斑块稳定性。
4.4 硫氧还原蛋白 Thioredoxin-1(Trx-1)是一种分子量为12 kD、高度保守的氧化应激抑制蛋白,具有抑炎、抗凋亡等作用[50]。Trx-1 能减少过氧化,下调IL-4 刺激的巨噬细胞中细胞周期蛋白依赖性激酶抑制剂p16INK4a 表达,或在LPS 诱导的巨噬细胞中降低AP-1 和Ref-1 表达,最终调节巨噬细胞向M2型极化。动物实验表明,Trx-1 与M2 型巨噬细胞共定位于动脉粥样硬化病变部位,显著减缓动脉粥样硬化病变,提示Trx-1 可通过调节巨噬细胞向M2 型极化延缓动脉粥样硬化发展[51]。近年Trx-1 的截短形式Trx-80对动脉粥样硬化的作用也有研究。Trx-80是由1~80 或1~84 个N-端氨基酸组成的C 端截短形式,具有细胞激酶样活性,被未知的酶在细胞表面切割,具有细胞因子样活性[52]。据报道,巨噬细胞会切割全长Trx-1 产生Trx-80。MAHMOOD 等[53]证实Trx-80 可促进M1 型巨噬细胞分化并促进动脉粥样硬化发展。
4.5 KCa3.1 通道 KCa3.1 即中间电导钙激活钾通道,是信号级联的一部分,在许多免疫细胞(包括T细胞、B细胞、小胶质细胞和巨噬细胞)活化、增殖、细胞因子分泌和体积调节过程中调控相对全局和长时间的钙升高[54]。RENDE 等[55]为探究KCa3.1通道在巨噬细胞极化中的作用及其与动脉粥样硬化斑块不稳定性的关系,采用联合结扎ApoE-/-小鼠左肾动脉和左颈总动脉的方法建立斑块不稳定模型,发现在人单核细胞向巨噬细胞分化过程中,KCa3.1表达显著上调,阻断KCa3.1可抑制STAT-1磷酸化,显著降低巨噬细胞极化过程中促炎基因表达,抑制巨噬细胞向M1型分化,动物模型中,KCa3.1阻断治疗可显著降低与M1 巨噬细胞极化相关标志物表达,增强动脉粥样硬化病变中M2 型标志物表达,降低颈动脉斑块破裂和管腔血栓发生率。
4.6 免疫蛋白酶体 26S 蛋白酶体复合物是UPS的蛋白水解中心,通常由3个催化亚基β1(PSMB6)、β2(PSMB7)和β5(PSMB5)组成,LMP10 是β2 受炎症刺激后的诱导形式[56]。研究报道,LMP10 缺失显著降低了主动脉巨噬细胞浸润,M2/M1 升高,同时促炎M1 细胞因子MCP-1、IL-1 和IL-6 表达降低,抗炎M2 细胞因子IL-4 和IL-10 表达增加,斑块坏死核心面积明显减少[57]。ox-LDL 诱导的体外泡沫细胞形成过程中,LMP10 缺失减弱了巨噬细胞极化和炎症,与IκBα降解和NF-κB激活降低有关。免疫蛋白酶体亚基LMP10 可能通过调节NF-κB 介导的巨噬细胞极化和炎症,促进饮食诱导的ApoE-/-小鼠动脉粥样硬化。靶向LMP10 可能是动脉粥样硬化的治疗方法。
4.7 HIF-1α/PDK4途径 最近研究发现,晚期糖基化终产物刺激巨噬细胞极化后会促进动脉粥样硬化发展。其中缺氧诱导因子-1α(hypoxia-inducible factor-1α,HIF-1α)和丙酮酸脱氢酶激酶4(pyruvate dehydrogenase kinase 4,PDK4)是调节葡萄糖代谢的两种关键蛋白。进一步研究发现,糖尿病ApoE-/-小鼠颈动脉中AGEs 表达和M1 巨噬细胞数量增加,此外,AGE-牛血清白蛋白通过HIF-1α/PDK4 轴诱导RAW264.7向M1型转化[10]。因此,靶向巨噬细胞的HIF-1α 或PDK4降低M1型巨噬细胞表达,可能有助于糖尿病动脉粥样硬化治疗。
4.8 其他 SIRT2 是一种NAD+依赖性脱乙酰酶,参 与 调 节 巨 噬 细 胞 极 化。ZHANG 等[58]用 雌 性LDLR-/-小鼠研究SIRT2 对动脉粥样硬化的作用,发现SIRT2 能抑制LDLR-/-小鼠动脉粥样硬化斑块进展和增强斑块稳定性,其作用与抑制巨噬细胞向M1型转化有关。
高密度脂蛋白(high-density lipoprotein,HDL)能够清除细胞中多余的胆固醇,对动脉粥样硬化起保护作用。SANSON等[59]用IL-4及HDL联合IL-4分别处理小鼠原代巨噬细胞,发现HDL、IL-4 共处理组M2 型标志物Arg-1 和Fizz-1 转录水平更高,iNOS、IFN-γ 转录水平降低。进一步提取STAT6-/-小鼠巨噬细胞检测各组转录水平,发现IL-4 或HDL 作用被抑制[58]。说明HDL 增加M2 型标志物表达、发挥抗炎作用需要STAT6参与。
肝 细 胞 生 长 因 子(hepatocyte growth factor,HGF)也称肝细胞生长素,广泛表达于各种组织和细胞,具有保护血管内皮细胞、抗炎、抗凋亡等作用。张亮等[60]用新西兰兔建立动脉粥样硬化模型研究HGF 对巨噬细胞表型及斑块稳定性的影响,发现与模型组相比,HGF 可抑制M1 型巨噬细胞浸润,诱导M2 型巨噬细胞分化,增加斑块胶原纤维和VSMCs含量,增强斑块稳定性,延缓动脉粥样硬化发展。但HFG 如何促进M1 型向M2 型转化的机制有待进一步研究。
5 小结
综上,巨噬细胞极化状态,尤其是M1/M2 比例对斑块的稳定性影响重大。研究已证实一定条件下,使M1 型巨噬细胞向M2 型巨噬细胞转换有利于提高斑块稳定性,但M2 型中不同亚型细胞对斑块的作用是否一致还不明确,相关机制也有待进一步研究。此外,巨噬细胞其他表型如M4、Mox、Mhem、M(Hb)型均与斑块稳定性相关(图1)。鉴于巨噬细胞的可塑性及其在动脉粥样硬化中的关键作用,以巨噬细胞表型调控为切入点或许能为动脉粥样硬化靶向治疗或新药研发提供新的思路。
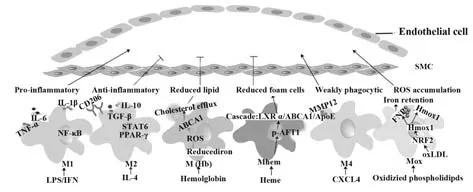
图1 巨噬细胞不同表型对斑块的作用Fig.1 Effects of different phenotypes of macrophages on plaque
猜你喜欢
现代财经-天津财经大学学报(2022年5期)2022-06-01
现代园艺(2017年21期)2018-01-03
电子测试(2017年15期)2017-12-18
中华老年多器官疾病杂志(2016年9期)2016-04-28
中国康复理论与实践(2015年10期)2015-12-24
电源技术(2015年1期)2015-08-22
医学研究杂志(2015年7期)2015-06-22
医学研究杂志(2015年5期)2015-06-10
现代检验医学杂志(2015年6期)2015-02-06
现代检验医学杂志(2015年5期)2015-02-06
