
核壳结构中手性分子诱导的等离激元光学活性
2023-11-08 12:13孙智广朱国栋方蔚瑞
辽宁石油化工大学学报 2023年5期
闫 荣, 孙智广, 高 楠, 朱国栋, 方蔚瑞
(大连理工大学 物理学院,辽宁 大连 116024)
贵金属纳米颗粒因其独特的性质被广泛关注,如何制备稳定性好、分散性好的纳米颗粒一直是纳米光学领域研究的热点。纳米颗粒的制备在过去的数十年间被不断改进,目前已经报道了各种各样的纳米结构[1-2],相应的光学特性也已经被深入研究[3-4]。1987 年,A.HENGLEIN 等[5]通过向CdS 胶体溶液中添加二氧化钛(TiO2)或氧化锌(ZnO),发现TiO2颗粒能够显著提高CdS 的光电性质。1988 年,H.C.YOUN 等[6]利用二十四烷基磷酸(DHP)囊泡制备并稳定半导体颗粒,通过吸收荧光光谱和X 射线衍射等手段发现这些半导体颗粒可以针对性地调节其组成和价带带隙,并高效地促进催化反应,表明非均质复合半导体纳米颗粒比单个颗粒具有更好的光电特性。由于核壳结构具有组成灵活、制备容易等优点,吸引了研究者的广泛关注。他们通过特定的方式将贵金属纳米颗粒进行包覆。结果表明,在保证核的稳定性的同时,由于壳层的存在,核的光学性质[7-8]、物理化学性质[9]会发生一定的改变。Stöber 方法[10]是利用碱加快硅酸酯类在醇溶液中水解浓缩,制备单分散的SiO2小球。该方法被广泛应用在对金属、金属氧化物的包覆,使结构在溶液中具有很好的分散性。 1996 年,L. M. LIZ -MARZAN 等[11]首次得到了Au 核SiO2壳的核壳结构:用硅烷偶联剂对Au 表面修饰带上硅羟基,然后利用硅酸盐水解与硅羟基反应生成SiO2壳层。但是,引入的硅烷偶联剂影响颗粒的稳定性。2002 年,Y.LU 等[12]利用异丙酮为溶剂合成了核壳结构。2003 年,C.GRAF 等[13]利用聚乙烯吡咯烷酮对各种纳米粒子修饰后再包覆SiO2,结果表明,在保证核壳结构的同时具有很好的稳定性。核壳结构目前被广泛应用于纳米光学领域。J.F.LI 等[8]制备了Au@Pd核壳结构,通过改变核和壳的厚度实现了对其光学性质的调谐;F.WANG 等[9]通过研究核壳结构中的能量转移,实现了上转换的调节。另一方面,手性广泛存在于自然界中,类似于人的左右手,互为手性的两个对映结构镜像对称,但不能通过平移旋转等方式重合。对手性性质的研究已经遍及科学研究的方方面面[14-15]。不同构型的手性药物对大部分生物而言,其生理活性或毒性都有很大的区别。20 世纪60年代,大规模的婴儿畸形事件就是由于药物“反应停”中S 构型分子具有强烈致畸作用导致的,正是这一事件引起了人们对手性分子的广泛关注[16]。为了避免药物的副作用,分子检测和分离对映体变得极为重要[17]。但是,手性分子的光学活性响应很弱,且大多数分子的共振波长位于100~450 nm 的紫外波段[18],在长时间探测过程中具有高能量的紫外曝光会损坏样品,只有当分子的浓度较大时才能检测到其圆二色性(CD)信号[19]。因此,增强分子的CD 信号并将其共振波长转移到可见光范围内,对生物研究和药品生产具有重要意义。研究表明,当手性分子吸附在金属纳米颗粒上时,金属纳米颗粒的等离激元特性可以显著增强分子的CD 信号,且在等离激元共振峰的位置处会产生新的CD 响应[20-24]。据文献[25]报道,当手性分子与非手性分子相连时,在手性分子的诱导下会在非手性分子的吸收峰出现新的CD 响应。N.SHUKLA 等[26]化学合成了Au 纳米颗粒,并在其表面吸附D-或L-半胱氨酸的手性分子,利用等离激元的特性将CD 信号诱导至可见光范围内,从而有利于分子的测量,为生命科学研究提供了新思路。
本文首先利用溶胶凝胶法制备了Au 核SiO2壳(Au@SiO2)壳层结构。由于核壳结构具有很好的溶液稳定性,将手性分子局域在壳层中间,形成了手性分子的核壳结构(Au@molecule@SiO2),利用分子和等离激元的相互作用,在可见光波段诱导产生新的CD 信号。在此基础上,利用Mie 理论和核壳杂化理论,计算模拟了在圆偏光入射条件下Au@molecule@SiO2的光学响应,同时研究了偶极子在诱导CD 中的贡献。
1 实验部分
1.1 试剂与仪器
试剂:氯金酸,分析纯,北京百灵威科技有限公司;柠檬酸三钠、硅酸钠,分析纯,天津市大茂化学试剂厂;L-/D-缬氨酸甲酯盐酸盐,分析纯,大赛璐药物手性技术(上海)有限公司。
仪器:MIRA3-LMH 扫描电子显微镜,TESCAN公司;J-810圆二色光谱仪,日本JASCO 公司。
1.2 实验方法
Au NPs 的合成方法(30 nm):向圆底烧瓶中加入20 mL 去离子水和60.0 μL(0.1 mol/L)氯金酸,在90 ℃的温度下加热搅拌;向溶液中加入1.4 mL柠檬酸三钠溶液,30 min 后溶液变红色表示种子生成;加入250.0 μL 柠檬酸三钠溶液,2 min 后继续加入60.0 μL 氯金酸并搅拌30 min,此时种子进行第一次生长;重复生长步骤2 次后,将溶液自然冷却至室温;用超纯水洗3 次(以10 000 r/min 的转速离心10 min)后分散在纯水中。
手性分子修饰Au NPs 合成方法:取5.0 mL Au NPs 原液分散在装有5.0 mL 超纯水的圆底烧瓶中,加入100.0 μL 手性分子乙醇溶液(0.1 mol/L),在室温条件下陈化3 h 后直接测量。
Au@molecule@SiO2合成方法[27]:将手性分子修饰过的颗粒水洗2 次(去掉溶液中多余的分子)后,将其分散到5.0 mL 的去离子水中。向溶液中加入3.2 mL 配置好的稀硅酸钠溶液(pH=10.3),室温搅拌3 min,将温度升高至90 ℃,反应3 h 后冷却到室温,水洗3 次后将其分散到5.0 mL 去离子水中。
采用扫描电子显微镜(SEM)观察结构的形貌,In-Beam SE 工作模式下工作电压为20 kV,工作距离为5 mm。通过圆二色光谱仪获得紫外可见(UV)吸收光谱和圆二色性(CD)光谱。
1.3 理论方法
通过Mie 理论能够快速准确地得到球形颗粒体系的解析解,广义的Mie 理论还可以用于计算球壳及多球体系[28]。通过麦克斯韦方程组求解球形结构散射场的解析解,其过程是将入射场和散射场表示为横电波M 和横磁波N 的展开式,并通过边界条件求解,求得散射场展开系数和入射场展开系数之间的关系[29]。得到解析解后,利用它计算相应的散射谱、吸收谱和近场的电磁场分布。对于核壳结构,从最内层开始利用边界条件推导出每层的散射系数an、bn,消光截面积Cext、吸收截面积Cabs和散射截面积Csca[30]:
式中:ψn、ξn、χn分别对应不同的Bessel 函数;mr为第r层的相对折射率;yr=kr Rr,kr为第r层的波数,Rr为第r层的半径均为系数,由边界条件决定。
Mie 理论中颗粒散射场可以被分解成一系列多极子辐射场的叠加,每种多极子由对应阶数的电磁多极散射系数(an、bn)来描述。
对于Au@molecule@SiO2结构,Au 的介电常数εAu来自文献[31],手性层的介电常数εc、手性参数ξc表达式为[24]:
式中:σCD为圆二色信号,mdeg;C为消光截面积;下标L 和R 分别对应左旋圆偏振光和右旋圆偏振光。
本文所有基于Mie 理论的计算均由自编代码计算完成。
2 结果与讨论
2.1 Au 和Au@SiO2的光学特性
图1 为Au NPs、Au@SiO2的SEM 和UV 光谱。

图1 Au NPs、Au@SiO2的SEM 和UV 光谱
从图1(a)可以看出,合成的Au NPs 粒径均一,平均粒径约为30 nm。从图1(b)可以看出,530 nm处有明显的吸收峰。从图1(c)和图1(d)可以看出,Au@SiO2核壳结构呈现出明显的SiO2壳层,壳层厚度约为10 nm,同时在536 nm 处有明显的吸收峰。与Au NPs 相比,由于高折射率SiO2壳层的存在,该核壳结构的吸收谱发生了略微红移现象。
2.2 Au 和Au@SiO2的手性响应
选取L-/D-缬氨酸甲酯盐酸盐,对Au NPs 进行了修饰。图2 为纯手性分子溶液、手性分子修饰的Au NPs 溶液、Au@molecule@SiO2的UV 光谱和CD光谱。

图2 纯手性分子溶液、手性分子修饰的Au NPs 溶液、Au@molecule@SiO2的UV 光谱和CD 光谱
从图2(a)和图2(b)可以看出,在紫外波段220 nm 处有明显的吸收峰;L-溶液的CD 光谱在220 nm处呈现一个正的科顿效应峰,而D-溶液则相反,这对应于互为手性的分子的CD 信号。在此基础上,将手性分子和Au NPs 溶液在室温条件下陈化搅拌3 h,在纳米颗粒充分吸附手性分子后直接进行了UV 光谱的测量。从图2(c)可以看出,吸附手性分子的Au NPs 的等离激元共振吸收在530 nm,相比于吸附分子前,它的峰值未发生明显的变化,但是在该体系中也没有明显的分子吸收峰(220 nm 处)。从图2(d)可以看出,L-/D-修饰的Au 颗粒溶液在220 nm 处出现具有科顿效应的手性分子CD 信号,未加分子修饰的Au NPs 并未出现手性分子CD 响应。此外,还可以观察到在Au 等离激元共振位置处出现的手性分子诱导CD 响应,这是由于手性分子和贵金属纳米颗粒之间的相互作用,导致Au NPs出现了手性特性。 从图2(e)可以看出,Au@molecule@SiO2的吸收峰位为538 nm,与Au 颗粒的共振峰(530 nm)相比发生了红移,与未吸附分子的结构(Au@SiO2)的红移现象保持一致,同时在220 nm 处看到较弱的分子吸收峰。从图2(f)可以看出,除了分子本身的CD 响应,在等离激元共振峰位置530 nm 处,存在互为镜像的2 个科顿效应峰,表明手性分子的加入使纳米颗粒产生了手性响应。此外,相比于未加入手性分子的Au@SiO2,加入手性分子的体系显示出2 nm 的红移,这主要是由于手性分子的吸附使局部电场受到影响,导致等离激元共振峰发生变化,此时诱导产生的CD 信号相比于直接在溶液状态下修饰Au NPs 更容易体现。这可能来自于两方面的原因:一是相对于直接分散的手性分子修饰的Au 颗粒,SiO2包裹的Au 颗粒周围的电磁场更有局域性,使分子与Au NPs 的作用更强;二是由于SiO2壳的存在,金核与壳相互作用,在壳内表面诱导出表面电荷,从而增强分子与Au 颗粒的相互作用。值得注意的是,由于手性分子直接修饰Au NPs 和Au@molecule@SiO2两种情况下加入的手性分子物质的量一致,导致分子在220 nm 处的CD 强度相同,此外核壳结构洗掉多余的手性分子,同时利用SiO2壳层将其保护起来,因此可见光波段诱导的CD 响应能够得到很好的表现。
为了进一步验证所合成的手性核壳结构具有优异的稳定性,考察了被手性分子修饰的Au NPs和Au@molecule@SiO2的吸光度随时间的变化情况,结果见图3。从图3 可以看出,随着时间的推移,手性分子修饰的Au NPs 的吸光度逐渐下降,同时在接近800 nm 处出现了新的峰,这表明在Au NPs 中存在手性分子时,它们会发生团聚,导致溶液的稳定性较差;Au@molecule@SiO2的吸光度随时间的变化较小,表明该溶液具有很好的稳定性。
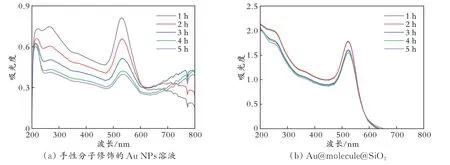
图3 手性分子修饰Au NPs 溶液和Au@molecule@SiO2的吸光度随时间的变化曲线
3 理论分析
采用Mie 理论对分子修饰的Au 单颗粒(R=15 nm)的吸收光谱和CD 光谱进行了数值模拟,手性分子的参数来自文献[32,34]。图4 为手性分子修饰的Au NPs 溶液和Au@molecule@SiO2的Mie 理论计算结果。从图4(a)可以看出,当没有手性分子存在时,220 nm 处没有吸收峰,但是当手性分子存在时,产生了强烈的吸收峰,相比实验结果(见图2(c)),吸收峰更为明显,且峰值和单独的手性分子(见图2(a))一致。从图4(b)可以看出,除了在220 nm 处存在手性分子的CD 信号,在Au NPs 的共振峰位置530 nm 出现了诱导CD 效应,相比实验结果(见图2(d)),同样具有手性分子信号,相比CD 光谱实验结果(见图2(d)),CD 信号较强。这是因为利用Mie 理论模拟计算的是单颗粒的响应,而实验测量的溶液状态中Au NPs 数量众多且尺寸有一定的分布,产生的CD 信号很容易被淹没。此外,构建一个三层结构来模拟实验合成的Au@molecule@SiO2。根据Au@SiO2结构的SEM 图(见图1(c)),无手性分子时,SiO2层厚度d=10 nm,存在手性分子时,手性分子层厚度d1=2 nm,SiO2层厚度d2=8 nm,整体结构大小一致。从图4(c)可以看出,核壳结构的吸收峰与实验结果相符(见图2(c))。从图4(d)可以看出,吸附分子的结构清晰地显示出分子本身的CD 信号,同时也显示出在等离激元共振峰位置(530 nm)的诱导CD 信号。与理论计算相比,实验测量(见图2(f))得到明显的CD 信号,由于SiO2壳层的存在阻止了分子的团聚,减弱了颗粒之间的相互作用,同时在溶液状态下众多颗粒信号叠加得到明显的CD 信号。对比两种情况计算得到的CD 光谱可知,当分子局域在核壳结构中时,颗粒表面的等离激元共振会受到壳层的屏蔽和干扰[35],从而导致产生的诱导CD 强度小于溶液状态下。

图4 手性分子修饰Au NPs 溶液和Au@molecule@SiO2的Mie 理论计算结果
为了进一步理解Au@molecule@SiO2中诱导的等离激元共振位置处的CD 信号机制,计算了手性核壳结构各个部分和整体结构的吸收CD 光谱及混合极化率,结果见图5。

图5 Au@molecule@SiO2结构中各个部分和整体结构的吸收CD 光谱及混合极化率
从图5(a)可以看出,Au 核在220 nm 处出现正信号,同时在等离激元共振位置出现诱导CD 信号,说明当分子存在时,由于分子与Au 核的相互作用导致Au 的圆偏光响应发生了变化;对没有手性层的Au 核不存在CD 贡献。从图5(b)可以看出,molecule@SiO2与Au 核的响应(见图5(a))信号正好相反,表明分子层与等离激元相互作用后,既可以诱导出Au 核上的CD 响应,同时等离激元也将手性传递到分子上,这是一个耦合的过程;没有Au 核存在时,手性壳层只有分子的CD 信号。从图5(c)可以看出,除了分子自己的CD 响应外,在紫外波段还存在诱导CD 信号,整体的CD 响应是由Au 核和手性分子层光学活性响应的场相干叠加的结果。由此进一步证实了由于Au 核和手性壳层之间存在相互作用,使整体结构具有CD 响应。对于小尺寸的手性核壳结构,电磁效应占主导地位,分析了Au核和手性壳层之间的电磁相互作用在CD 信号中的贡献,图5(c)中的蓝色实线与红色实线基本吻合,揭示了核壳结构下手性分子诱导的CD 主要来自电偶磁偶的相互作用[24,35]。
为了研究核壳结构中电偶极子和磁偶极子在散射CD 中的贡献,计算了手性核壳结构下各部分电偶极子和磁偶极子的贡献,结果见图6。从图6(a)和图6(b)可以看出,分子诱导均在220 nm 处存在CD 响应,但是磁偶极子的贡献相比于电偶极子小103,几乎可以忽略;单独的Au 颗粒不存在电偶极子和磁偶极子贡献。从图6(c)可以看出,在530 nm 处出现强度很大的诱导CD 信号,表明分子壳层中的偶极子占主要贡献。从图6(d)可以看出,除了分子的CD 外,并不存在诱导CD 的贡献。对比图6(a)、(b)、(c)和(d)可知,单独的Au 核和手性壳层并没有产生诱导CD,诱导CD 信号来自Au 核和手性壳层之间的作用。从图6(e)可以看出,整体和图5(c)一致,但是等离激元共振位置处的强度很小,说明散射CD 贡献相比于吸收可以忽略。从图6(f)可以看出,只存在分子的CD,对诱导CD 并没有贡献。综上所述,电偶极子和磁偶极子散射CD 相比于吸收更小,几乎可以忽略,也进一步说明了整个结构吸收CD 占主要贡献。

图6 Au@molecule@SiO2结构中各部分电偶极子和磁偶极子的散射CD 谱
4 结 论
通过液相还原法成功合成了带有手性分子的Au@SiO2核壳结构,这种结构相比于分子直接修饰的Au NPs 具有良好的稳定性。L-/D-缬氨酸甲酯盐酸盐作为修饰分子,在220 nm 处存在强烈的吸收及CD 响应。当分子直接修饰Au 颗粒时,溶液状态下除了分子的CD 外,分子和金属等离激元的相互作用诱导产生可见光波段CD并未被检测到。 将分子局域在核壳结构(Au@molecule@SiO2)中后,可见光波段的诱导CD 信号被明显检测。利用Mie 理论对实验结果计算模拟,对比后发现与实验结果有很好的一致性。同时,利用多级展开方法,揭示了结构中的诱导CD 主要来自电偶极子和磁偶极子的相互作用,也进一步分析了电偶极子和磁偶极子在散射CD中对整体结构的贡献,结果表明电偶极子在核壳结构散射中占主要地位。合成的带有手性分子的核壳结构很好地保护了分子,为今后分子的应用提供了新的方式,对金属核壳结构的应用具有重要参考意义。
猜你喜欢
分子催化(2022年1期)2022-11-02
原子与分子物理学报(2021年2期)2021-03-29
农村青少年科学探究(2020年5期)2020-08-18
新民周刊(2018年8期)2018-03-02
饮食科学(2017年12期)2018-01-02
西安工程大学学报(2016年6期)2017-01-15
材料科学与工程学报(2016年1期)2017-01-15
国外医药(抗生素分册)(2016年4期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
湖北师范大学学报(自然科学版)(2015年1期)2016-01-10
