
16α-羟基泼尼松龙合成方法综述
2023-11-07 11:52:58蒋建武江海尹琳琳林建东
化工生产与技术 2023年5期
蒋建武,江海,尹琳琳,林建东
(台州仙琚药业有限公司,浙江 台州317016)
16α-羟基泼尼松龙(1),英文名称为16α-hydroxprednisolone,分子式为C21H28O6,化学名称为11β,16α,17α,21-四羟基孕甾-1,4-二烯-3,20-二酮,是一种重要的甾体医药中间体,可用于制备布地奈德(2)、环索奈德(3)和地索奈德(4)等糖皮质激素药物[1-3]:



糖皮质激素类药物广泛应用于顽固性哮喘、慢性阻塞性肺病等炎症性呼吸道疾病的治疗。其中布地奈德和环索奈德,具有使用剂量小、局部抗炎作用强和不良反应少等优点,不仅是儿童雾化吸入常用药物,还是临床治疗严重性哮喘和过敏性鼻炎的首选药物[4-8]。此外,奈德类糖皮质激素类药物在新型冠状病毒感染的治疗中,能够快速抑制炎症反应,有效缓解初始症状[9]。
16α-羟基泼尼松龙作为合成奈德类糖皮质激素药物的基础原料,市场应用前景十分广阔。制备方法主要包括化学合成法和微生物发酵法。化学合成法采用泼尼松龙或醋酸泼尼松为起始原料,普遍存在消除反应选择性差、总收率低和工艺三废量大等缺点,且中间体邻二醇的制备不可避免采用重金属锰试剂,不利于工业化生产。微生物发酵法主要以泼尼松龙为起始原料,通过微生物发酵技术直接生成16α-羟基泼尼松龙。生物催化法具有步骤少、选择性好等优点,研究人员已经在该领域做了初步研究。然而,现有菌种活性差,发酵液中有效物含量低、提取成本较高,制约了生物催化法在16α-羟基泼尼松龙工业化制备中的进一步应用。
1 以泼尼松龙为原料
KIM 等最早提出了以泼尼松龙(5)为起始原料制备16α-羟基泼尼松龙的方法[10](Ac 为乙酸根,Py为吡啶,DMF为N,N´-二甲基甲酰胺):
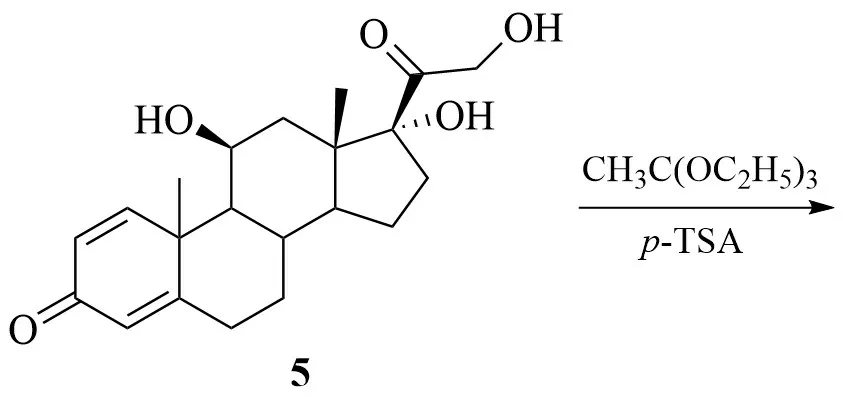

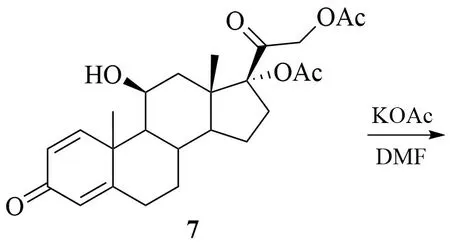

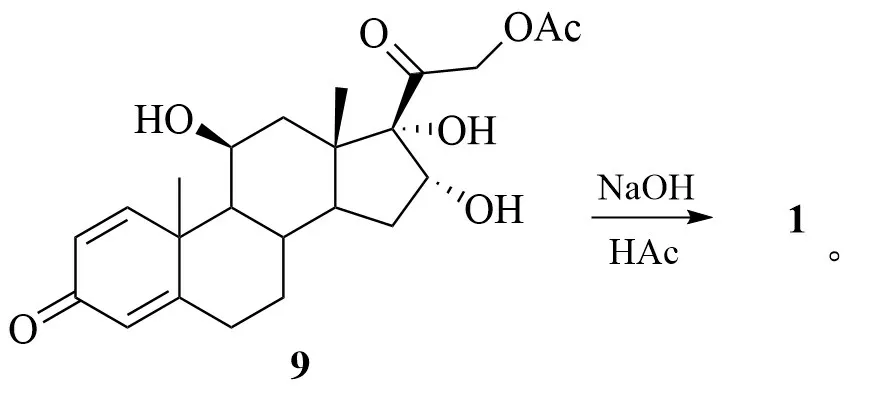
5 在对甲苯磺酸(p-TsA)的催化作用下,与原乙酸三乙酯(CH3(OC2H5)3)经酯交换反应得到化合物6;6 再经水解、酰化等步骤得到17α-脱羟醋酸泼尼松龙(7);在碱性条件下,7 经消除C-17位乙酰基,得到不饱和烯酮结构化合物8;8 经高锰酸钾(KMnO4)双键氧化反应得到邻二醇化合物9;9再经水解反应得到目标产物1。
该方法路线较为繁琐,所用试剂对环境污染较大,总收率较低仅为4.9%,不适用于工业化生产。
殷超等对上述反应路线进行了改进,将生产工艺由6步反应缩短至4步[11]:
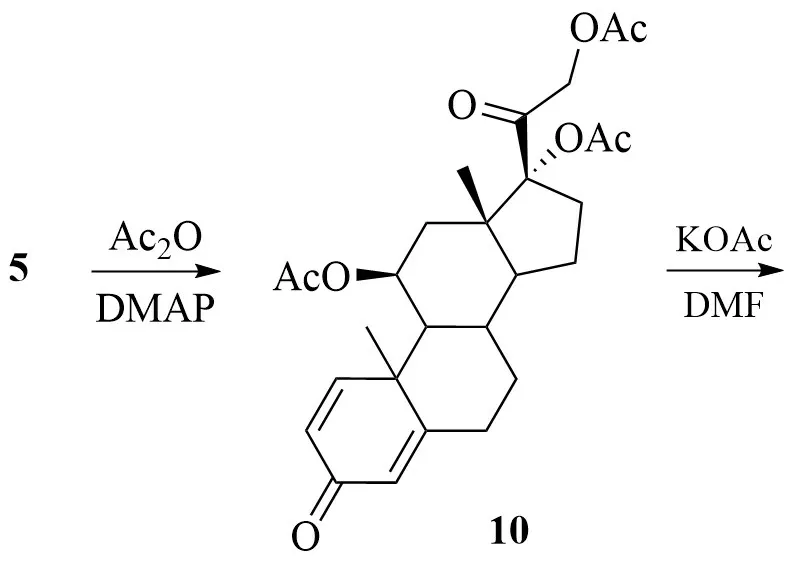
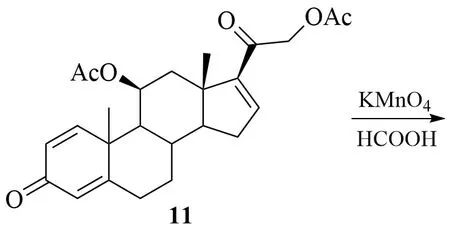

5 直接在对二甲氨基吡啶(DMAP)催化下,通过与醋酐的酰化反应保护C-11、C-17、C-21 位处羟基结构,得到三醋酸酯中间体化合物10;10经消除、氧化及水解等步骤制得目标产物1。
该方法产品收率高,生产成本降低。但是,中间体的制备仍需使用KMnO4,导致产品重金属残留较高,三废处理难度大。
裴文等以酸性离子液体作溶剂及脱水反应催化剂,双氧水(H2O2)作氧化剂,实现了16α-羟基泼尼松龙的合成[12]:


5在咪唑类酸性离子液体中,先进行脱水反应得到烯酮中间体13;13经氧化得到目标产物1。
该方法具有创新性,使用离子液体作为反应介质和催化剂,路线设计简单,总收率较高约85%。但是,离子液体造价昂贵,核心步骤反应时间较长,且过量的H2O2使用存在较高风险,不利于工业化生产。
2 以醋酸泼尼松为原料
王勇报道了1 种以醋酸泼尼松(14)为起始原料制备16α-羟基泼尼松龙的方法[13]:



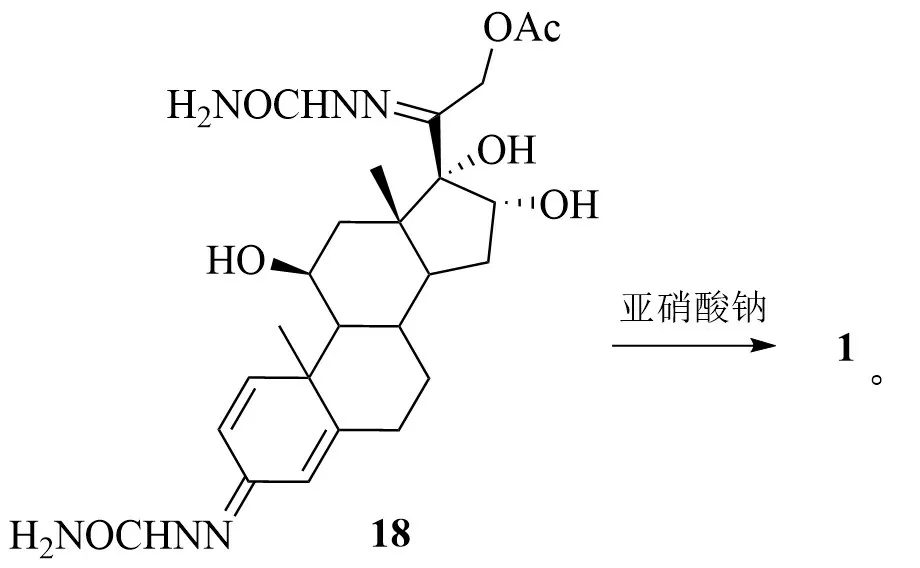
14 经消除反应脱去C-17 位羟基得到不饱和烯酮中间体化合物15;15 经KMnO4氧化得到邻二醇化合物16;16 与氨基脲(NH2NHCONH2)通过缩合实现C-3 和C-20 酮基的选择性保护得到化合物17;17 被硼氢化钾(KBH4)选择性还原得到三羟基化合物18;18经水解反应制得目标产物1。
该方法通过加入阻滞剂抑制KMnO4对反应物的氧化速度,提高了氧化反应选择性,但需要多步保护和脱保护反应,反应步骤长,原子经济性低,成本较高。
杨坤等报道了1 种以14 为起始原料制备16α-羟基泼尼松龙的新方法[14]:
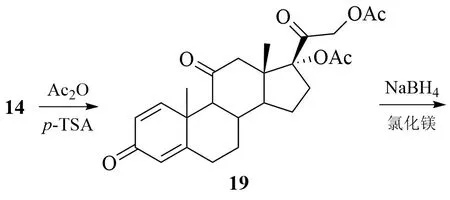

14首先在p-TsA催化下,经醋酐的酰化反应得到双醋酸酯化合物19;19 的C-11 位酮基经硼氢化钠(NaBH4)选择性还原得到化合物7;7 经消除、氧化及水解反应制得目标产物1。
该方法虽然可以避免多步保护和脱保护反应,但也存在锰试剂使用量大和还原反应选择性差等不足。
甘红星等报道了1 种以14 为起始原料,通过制备关键中间体17α-脱羟醋酸泼尼松(8)来合成16α-羟基泼尼松龙的方法[15]:




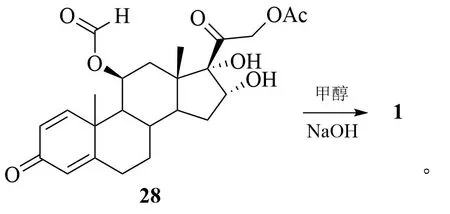
14 首先与三甲基氯硅烷(TMSCl)在有机碱催化下发生C-11 位硅醚化反应得到化合物21;21在三氧化硫/Py体系中发生C-17位脱水反应,并在酸性条件下进一步水解脱硅醚保护得到8;8 与间氯过氧苯甲酸(m-CPBA)发生环氧化反应得到化合物22;22 与冰醋酸在对甲苯磺酸催化下开环得到化合物23;最后,23 在氧化铝固相碱催化剂的催化下水解得到目标产物1。
该方法克服了传统消除工艺副反应多和氧化工艺需采用重金属锰等缺点,工艺更绿色、清洁,降低了生产成本。
3 其他甾体化合物作为原料
近年来,研究人员也报道了采用其他甾体化合物为原料合成16α-羟基泼尼松龙的方法[16-18]:

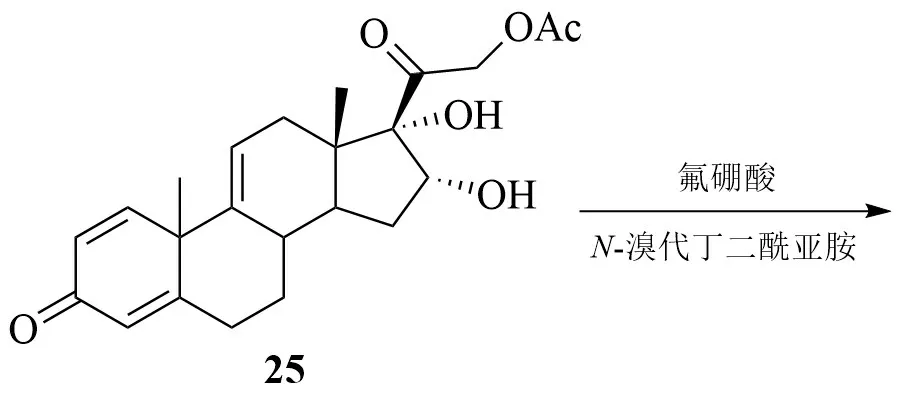


李文宽等以21-羟基孕甾-1,4,9(11),16(17)-四烯-3,20-二酮醋酸酯(24)为原料,首先经KMnO4氧化得到16,17,21-三羟基孕甾-1,4,9-三烯-3,20-二酮-11-甲酸酯-21-醋酸酯(25);25经双键溴羟化反应得到溴羟物26;以锌粉/氯化铬为还原体系,巯基乙酸(TGA)为供氢剂,26 可经还原脱溴得到9;9经水解得到目标产物1。
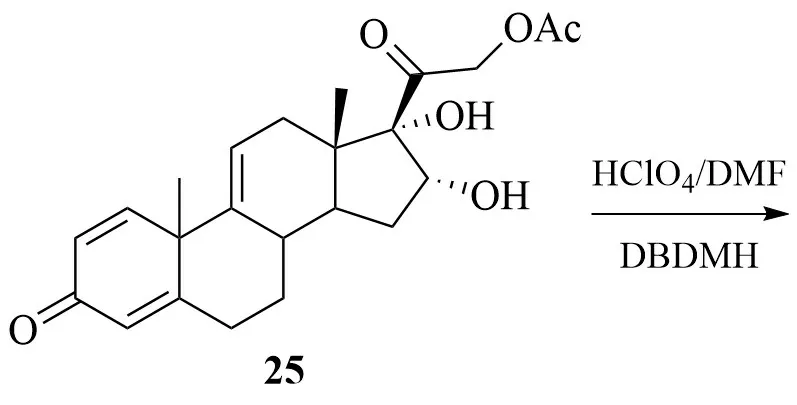

邵振平等则直接以25 为原料,首先在高氯酸(HClO4)、DMF、二溴海因(DBDMH)体系中进行双键加成反应得到溴代甲酸酯中间体化合物27;27经还原脱溴及水解得到目标产物1。
虽然采用上述甾体化合物作为起始原料可以缩短反应步骤,提高路线总收率,但是关键步骤仍需采用金属铬等剧毒试剂,工业化应用受限。
4 微生物发酵法
以廉价易得的甾体化合物为原料,采用生物发酵技术直接制备16α-羟基泼尼松龙是当下的研究热点,研究人员已在该领域作了初步研究。例如,张文权等采用玫瑰产色链霉菌菌株TS-58发酵得到16α-羟基泼尼松龙,泼尼松龙转化率为13.8%[19]。吴倩等利用双氧水代替天然的电子传递系统,实现了玫瑰产色链霉菌P450s酶体外催化泼尼松龙得到16α-羟基泼尼松龙,泼尼松龙转化率达到了35.7%[20]。RESTAINO 等将单纯节杆菌和玫瑰产色链霉菌进行全细胞偶联,氢化可的松经生物转化直接得到16α-羟基泼尼松龙,收率达到68.8%[21]。
5 结束语
综上所述,16α-羟基泼尼松龙有4 种合成方法。化学合成法主要采用泼尼松龙或醋酸泼尼松为起始原料,经5步左右化学反应制得,但普遍存在消除反应选择性差、总收率低、工艺三废量大等缺点,且中间体邻二醇的制备不可避免需采用重金属锰试剂。虽然也发展了一些更绿色的邻二醇制备方法,但收率及工艺成本尚未达到工业化要求。另外,泼尼松龙或醋酸泼尼松的成本较高,研究人员尝试了将其他甾体化合物作为原料,但仍存在关键步骤选择性较差、收率低及路线设计不够绿色环保等缺点[19-21]。相比化学合成法,生物发酵法具有步骤少、选择性好等优点,研究人员已经在该领域做了初步研究,然而现有的菌种活性差,发酵液中16α-羟基泼尼松龙的有效物含量低,提取成本较高,上述缺点制约了生物催化法在16α-羟基泼尼松龙工业化制备中的进一步应用。
展望未来,建议16α-羟基泼尼松龙的工艺改进方向为:
1)过硫酸盐参与的烯烃顺式双羟化反应已获得了一定的进展,未来有望应用于16 位双键氧化,实现对KMnO4氧化工艺的替代[22]。
2)光催化技术应用于还原脱溴反应最近已成功开发,后期结合较成熟的光催化放大反应器可应用于还原脱溴工艺,从而在工艺源头避免金属铬的使用[23]。
3)随着合成生物学的不断发展,16α-羟基化生物发酵技术一旦获得突破,将颠覆现有合成工艺。此外,若能以更大宗廉价的甾体化合物作为起始原料,通过生物发酵降解技术制备16α-羟基泼尼松龙或其相关中间体,将大幅度降低现有的工艺成本,实现制备工艺的变革。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
云南化工(2021年11期)2022-01-12 06:06:20
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
中华养生保健(2020年1期)2020-11-16 00:47:44
广州化工(2020年8期)2020-05-19 06:23:56
华东理工大学学报(自然科学版)(2015年5期)2015-02-27 13:49:56
华东理工大学学报(自然科学版)(2015年5期)2015-02-27 13:49:56
中国药业(2014年24期)2014-05-26 09:00:14
中国医学科学院学报(2014年6期)2014-03-11 20:26:18
