
超高效液相色谱-串联质谱法同时检测肉制品中17 种合成色素的含量
2023-11-06 06:42彭青枝周陶鸿王福燕
食品工业科技 2023年21期
龚 蕾,韩 智,彭青枝,周陶鸿,黄 徽,于 果,王福燕
(1.湖北省食品质量安全监督检验研究院,湖北武汉 430075;2.国家市场监管重点实验室(动物源性食品中重点化学危害物检测技术),湖北武汉 430075;3.湖北省食品质量安全检测工程技术研究中心,湖北武汉 430075;4.江汉大学医学部,湖北武汉 430056)
食品色泽是食品品质的一项重要指标,艳丽的色泽可提高人们的消费欲望。合成色素作为食品添加剂广泛应用在食品工业中,具有稳定性高、着色能力强、成本低廉、易调色等优点[1]。但合成色素主要以甲苯、苯等为原料,经过磺化、偶氮化等化学反应而制得,经人体代谢分解,可生成潜在的致癌物质,损害身体健康[2]。
在肉制品工业中,合成色素的使用有着严格要求,我国仅允许胭脂虫红、诱惑红等色素在肉制品中限量、限范围使用,而柠檬黄、苋菜红等常见合成色素禁止在肉制品中使用[3-4]。但在利益的驱动下,一些不法商家存在超范围、超限量使用色素的现象,甚至添加非食用色素,达到改善肉制品色泽的目的,危害食品安全[5]。据报道,在市场监督管理部门的日常监督抽检和专项抽检中,发现肉制品中检出酸性橙Ⅱ(酸性橙7)、酸性红1 等非食用色素[6]。GB 5009.35-2016[7]、GB/T 9695.6-2008[8]、SN/T 1743-2006[9]及SN/T 3536-2013[10]等国家及行业标准中,规范了对柠檬黄、胭脂红、日落黄、酸性橙7 等色素的检测方法,但是针对肉制品中可能非法添加酸性红1、偶氮玉红等化合物还没有相应的检测标准方法,缺少同时检测多种色素的技术,日常监督检测存在一定盲区。
目前,检测肉制品中色素常用方法包括薄层色谱法[11]、高效液相色谱法[12-14]以及液相色谱-串联质谱法[15-18]等。蒋荣华等[19]采用高效液相色谱法对肉制品中的苋菜红、柠檬黄、胭脂红等6 种色素进行检测,方法的检出限为0.12~0.21 mg/kg,方法的回收率为70.33%~93.08%。王韦达等[20]建立了固相萃取-超高效液相色谱分析方法,对不同食品中的红2G 进行检测,方法检出限为0.03 mg/kg,平均回收率在80%以上。李道霞等[21]建立了阴离子固相萃取-HPLC 法测定肉制品中酸性橙Ⅱ、喹啉黄、红2G 等色素的方法,该方法定量限为0.6~1.5 mg/kg,平均回收率为65.2%~102.0%。高效液相色谱法是目前国家标准、行业标准检测色素的主要方法,但是存在定性不准、灵敏度较差等问题,相较于高效液相色谱和薄层色谱法而言,高效液相色谱-串联质谱法具有定性准确、分析时间短等优势。因此,本文针对肉制品中超量超范围以及非法添加色素的现象,通过对样品前处理及液相色谱-串联质谱法测定条件进行优化,以期建立一种快速同时测定肉制品中酸性红1、酸性橙7 等17 种合成色素含量的方法,为肉制品中合成色素的滥用提供一种高效便捷的快速筛查及定量仪器方法,为食品安全监管提供技术支撑。
1 材料与方法
1.1 材料与仪器
26 批次火腿肠、26 批次香肠、10 批次鸭脖、10 批次腊肉等样品 购自武汉市、鄂州市、黄石市的超市以及个体工商户;乙腈、正己烷、甲酸、氨水、乙酸铵 色谱纯,德国Merck 公司;硫酸铵 分析纯,国药集团化学试剂有限公司 ;标准品信息见表1。WAX 固相萃取柱(200 mg, 6 mL)、ACQUITY UPLC HSS T3 色谱柱(100 mm×2.1 mm,1.8 μm)美国Waters 公司;AB 4500 液质联用仪,配Multi-Quant 数据分析软件 美国AB SCIEX 公司;ME204电子天平 梅特勒-托利多仪器有限公司;MultiVortex 涡旋混合器 广州得泰仪器科技有限公司;CLT55离心机 湖南湘仪实验室仪器开发有限公司;0.22 μm水系滤膜 天津津腾公司;Milli-Q 超纯水系统 美国Millipore 公司。

表1 17 种色素标准物质信息Table 1 Information of seventeen colorants
1.2 实验方法
1.2.1 标准溶液配制 分别精密称取17 种待测成分的标准品适量于10 mL 容量瓶中(精确至0.0001 g),加入少量超纯水进行溶解,定容到刻度,获得不同化合物的单一标准储备溶液,标准储备液浓度范围为0.10~2.3 mg/mL,精密量取适量标准储备液,用水稀释,配制成浓度均为10 μg/mL 的混合标准溶液。所有标准溶液在4 ℃条件下贮存。
1.2.2 样品前处理 提取:取有代表性样品500 g,采用粉碎机粉碎后,再将其研磨成细粉状,装入洁净的玻璃盛样容器内,密封并标明标记。称取2.0 g(精确至0.01 g)粉碎样品于50 mL 聚丙烯离心管中,加入10 mL 水,涡旋混匀1 min,50 ℃下超声提取10 min,加入0.5 mL 饱和硫酸铵溶液,振荡混匀1 min,8000 r/min 离心10 min,转移上清液至25 mL具塞试管中,加入10 mL 水再提取一次,合并两次滤液,用水定容到25 mL,此为提取液。
净化:将25 mL 提取液全部转移到50 mL 离心管,加入正己烷20 mL,涡旋混匀2 min,8000 r/min离心5 min,弃去全部上层正己烷,取10 mL 下层溶液待净化。将10 mL 待净化液通过已活化的WAX固相萃取柱,分别用3 mL 水、3 mL 2%甲酸-甲醇淋洗,弃去淋洗液,用6 mL 5%氨水-甲醇洗脱,收集洗脱液,50 ℃水浴下氮吹至干,用水定容到1.0 mL,涡旋混匀,过0.22 μm 水系滤膜(聚醚砜材质),待上机。
1.2.3 色谱条件 色谱柱:ACQUITY UPLC HSS T3 色谱柱(100 mm×2.1 mm,1.8 μm);进样量:5 μL;柱温:35 ℃;流动相:A 相为5 mmol/L 乙酸铵,B 相为乙腈,流速0.3 mL/min;梯度洗脱条件如表2。

表2 梯度洗脱程序Table 2 The gradient elution program
1.2.4 质谱条件 离子源:电喷雾离子源;扫描方式:负离子扫描;检测方式:多反应监测;电喷雾电压:-4500 V;离子源温度:500 ℃;气帘气压力:35 psi;碰撞气压力:8 psi;雾化器压力:55 psi;辅助气压力:55 psi;入口电压:-10.0 V;碰撞室出口电压:-10.0 V。各色素的质谱参考条件见表3。
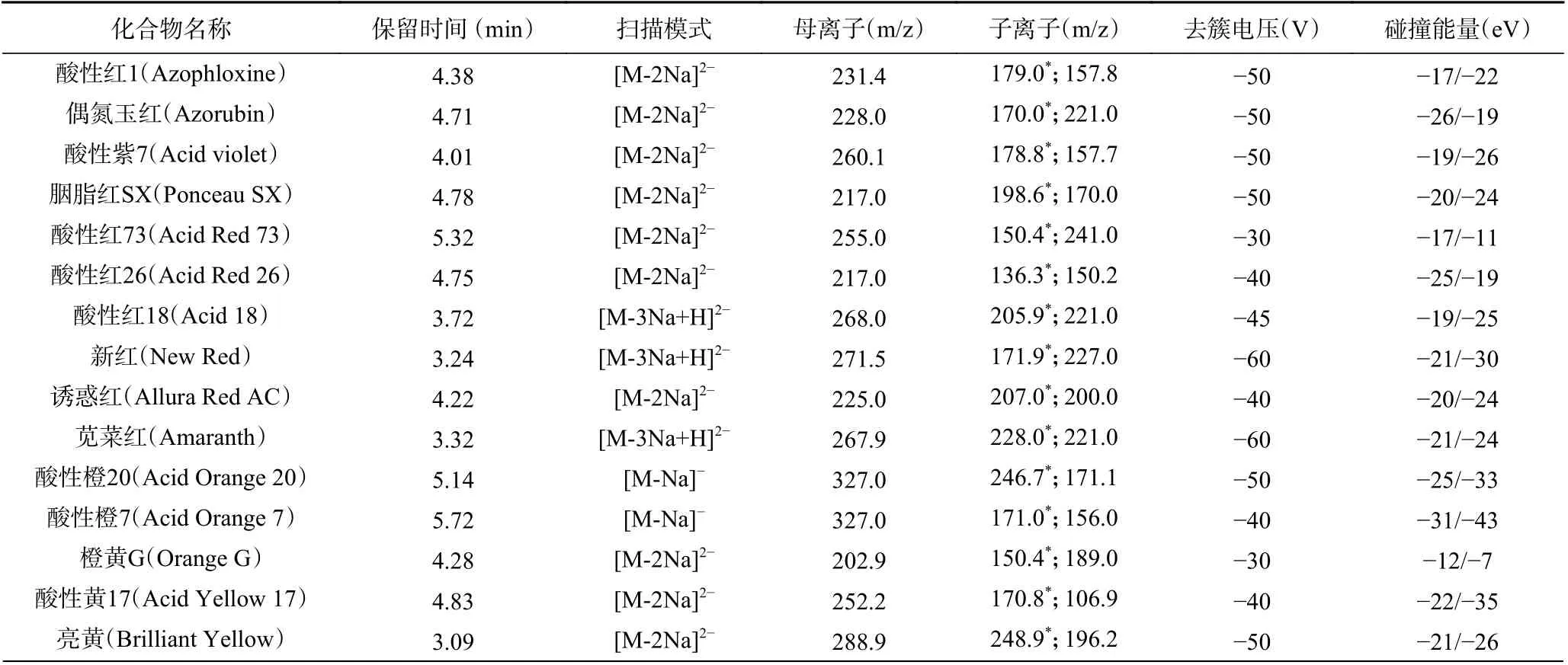
表3 17 种化合物的定性离子、定量离子、去簇电压和碰撞能量Table 3 Qualitative ion, quantitative ion, cone voltage and collision energy of seventeen compounds
1.3 数据处理
使用Analyst 软件采集数据,运用MultiQuant 3.0.2 进行定性定量分析,使用Origin 9.0 软件作图。
2 结果与分析
2.1 色谱条件与质谱条件的优化
为准确获得17 种色素的母离子,对质量浓度为0.5 mg/L 的各化合物单一标准溶液依次进行了一级质谱扫描。实验结果表明,在负离子扫描模式下,含有1 个钠离子的化合物能稳定形成[M-Na]-离子峰,含有2 个钠离子的化合物能稳定形成[M-2Na]2-离子峰,含3 个钠离子的化合物能同时稳定形成[M-3Na+H]2-和[M-3Na]3-离子峰,然而在后续的上机测试中,发现[M-3Na]3-响应极低,可能是因为[M-3Na]3-为源内裂解而产生[22],而[M-3Na+H]2-具有响应高且稳定的特点,因此选择[M-3Na+H]2-。为得到稳定性好、响应值高的定性定量子离子,对质谱和液相参数进行了优化,最佳参数见表3。在17 种化合物中,一共含有3 对同分异构体,分别是胭脂红SX 和酸性红26(分子式均为C18H14N2Na2O7S2),酸性红18 和苋菜红(分子式均为C20H11N2Na3O10S3),酸性橙7 和酸性橙20(分子式均为C16H11N2NaO4S)。在实际的色谱分离中,通过改变流动相的梯度,能够很好地将酸性红18 和苋菜红、酸性橙7 和酸性橙20 两对同分异构体进行色谱分离,再通过单一标准溶液确定其各自保留时间,苋菜红和酸性红18 的保留时间分别为3.32 min 和3.72 min,酸性橙20 和酸性橙7 的保留时间分别为5.14 min 和5.72 min,它们互不干扰,保留时间的不一致使得它们满足定性定量要求;改变流动相洗脱比例,很难将胭脂红SX 和酸性红26 分离,但它们碎裂的离子对具有独立性和专一性,产生的碎裂离子互不干扰,因此定性定量不受影响。酸性红18 和苋菜红,酸性橙7 和酸性橙20 的定量离子质谱图及总离子流图见图1。

图1 总离子流图Fig.1 TIC of all compounds
2.2 样品前处理的优化
2.2.1 净化与上机测试
2.2.1.1 固相萃取柱的选择 肉制品需通过净化以除去油脂等干扰物,保证上机样品不污染仪器。有研究通过稀释提取液100 倍来达到净化和减弱基质效应[17]。而更多的研究选择使用固相萃取柱对色素进行净化富集,包括WAX[23]、C18[24-25]、HLB[26],但尚无文献使用固相萃取柱富集净化的方法来同时测定文中所选取的17 种色素,因此本研究通过空白加标的方式验证了几款不同的固相萃取柱对这些色素的回收率,其中上样、淋洗、洗脱步骤按照萃取柱产品推荐的步骤进行。实验结果表明,只有WAX 同时对这17 种化合物具有较强的保留能力,回收率在70%~110%之间,满足实验需要,因此选择WAX 固相萃取柱。
2.2.1.2 上样溶液酸碱性的影响 pH 可以改变化合物的离子化或质子化程度,从而影响目标化合物在固相萃取小柱中的保留情况[27]。本实验比较了不同酸碱性下17 种化合物在固相萃取小柱的保留情况。将含量均为1000 ng 的17 种目标化合物分别添加到5 mL 水、5 mL 0.1 mol/L 硫酸铵、5 mL 1%氨水中,再全部通过已活化的WAX 固相萃取柱,收集全部流出液,并测试目标化合物含量。实验发现水和0.1 mol/L 硫酸铵上样后,流出液均不含目标物成分,说明WAX 固相萃取柱能很好地保留这17 种化合物。而1%氨水作为提取试剂进行上样,流出液含有部分目标物,特别是酸性红73、酸性红18、亮黄,在碱性上样条件下,目标物回收率不足50%。结合文献报道[28-29],在pH6.0 左右时,含有磺酸基的色素能在弱阴离子固相萃取柱上柱有最佳保留[30],因此本研究通过加入适量饱和硫酸铵溶液使上样呈弱酸性(pH6.0)来增加其在阴离子固相萃取柱上的保留。
2.2.1.3 淋洗溶液的选择 淋洗剂的选择是影响固相萃取柱回收率的重要因素,它不仅能去除非特异性结合的杂质,而且还能最大程度地保留目标化合物,从而提高检测的准确性和灵敏度[31-32]。选择5%甲醇-水、50%甲醇水、及5%甲醇-水(含2%甲酸)、50%甲醇水(含2%甲酸)、100%甲醇(含2%甲酸)进行淋洗,结果表明它们均能很好地保留目标化合物而不流出固相萃取柱,但使用100%甲醇(含2%甲酸)淋洗效果最佳,原因是此时能有效除去固相萃取柱残留的水分,下一步洗脱液在相同氮吹条件下能短时间内完全氮吹至干,有助于定容操作。
2.2.1.4 滤膜的影响 实验发现使用尼龙材质的滤膜,对17 种色素有不同程度的截留吸附,其中,通过尼龙滤膜后,偶氮玉红、胭脂红SX、酸性红73、酸性橙20、酸性橙7、亮黄被吸附100%,其他被吸附50%以上,而聚醚砜(PES)滤膜不吸附目标物,因此在进行过膜上机时,应严格测试所使用的滤膜,以保证合成色素不被滤膜材质所吸附。
2.3 线性范围、检出限、定量限、加标回收率及相对标准偏差
配制一系列标准溶液,以色素的质量浓度为横坐标,质谱的定量峰面积为纵坐标,绘制标准曲线,得出17 种色素在20~1200 μg/L 范围内具有良好的线性关系,R>0.99,以信噪比S/N=3 确定检出限,以S/N=10 确定定量限,17 种化合物检出限(LOD)为0.01~0.05 mg/kg,定量限(LOQ)为0.03~0.15 mg/kg。在香肠空白基质中分别添加不同浓度标准溶液,按照本文方法进行处理,按高中低三水平对空白样品提取液进行添加,每个添加水平平行测定6 次,计算回收率及相对标准偏差。各化合物在3 个添加水平(0.15、0.30 和1.0 mg/kg)下的回收率为71.2%~103.5%,相对标准偏差(RSDs,n=6)为1.3%~4.9%。线性范围、检出限、定量限、加标回收率及相对标准偏差具体结果见表4。

表4 17 种化合物的加标回收率、相对标准偏差、检出限、定量限及标准曲线及相关系数Table 4 Average spiked recoveries, relative standard deviations, LODs , LOQs, standard curve and correlation coefficient of seventeen compounds
2.4 实际样品检测结果
采用优化建立的UPLC-MS/MS 方法对市售香肠、火腿肠、熟制鸭脖、腊肉等72 份样品进行检测。结果显示检出诱惑红样品6 个,均为火腿肠,含量为0.71~2.7 mg/kg,含量低于国家标准限量15 mg/kg,为合格食品;检出酸性红1 样品4 个,均为香肠,含量为0.39~3.2 mg/kg,酸性红1 为非法添加色素,值得监管部门的关注。
3 结论
本文建立了同时测定肉制品中17 种人工合成色素的UPLC-MS/MS 检测方法。该方法重现性好、灵敏度高。17 种色素在20~1200 μg/L 范围内线性关系良好,R>0.99,方法的检出限为0.01~0.05 mg/kg,定量限为0.03~0.15 mg/kg。各色素在3 个添加水平(0.15、0.30 和1.0 mg/kg)下的回收率为71.2%~103.5%,相对标准偏差(RSDs,n=6)为1.3%~4.9%。对实际样品进行检测,结果表明在肉制品中含有非食用色素酸性红1,含量为0.39~3.2 mg/kg,值得监管部门的关注。本方法可实现同时分析肉制品中的17 种色素,大大提高了检验效率,为检验人员提供了一个简单、快捷、高效的检验方法,也为食品安全提供了技术保障,还可对肉制品中色素相关检测数据进行累积从而进行必要的风险评估,为政府监管部门提供数据支持,具有实际意义和一定社会效益。
猜你喜欢
云南化工(2021年10期)2021-12-21
今日农业(2020年23期)2020-12-31
中国油脂(2020年3期)2020-04-10
福建基础教育研究(2019年8期)2019-05-28
中学生数理化·八年级物理人教版(2017年6期)2017-11-09
无机化学学报(2016年8期)2016-12-06
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
化学分析计量(2016年1期)2016-03-14
分析测试学报(2015年3期)2016-01-13
食品工业科技(2014年11期)2014-03-11
