
骨髓间充质干细胞衰老与骨质疏松症的研究进展*
2023-11-02 10:14李小云王昊宇欧阳欣辰朱晓峰张荣华
中国病理生理杂志 2023年10期
李小云, 林 青, 王昊宇, 欧阳欣辰, 杨 丽, 朱晓峰,3,4, 张荣华△
(1暨南大学药学院,广东 广州 510632;2暨南大学中医学院,广东 广州 510632;3暨南大学第一附属医院,广东 广州 510630;4广东省中医药信息化重点实验室,广东 广州 510632)
骨质疏松症(osteoporosis, OP)是临床上常见的老年病,以骨量减少、骨微结构退化为特征,导致骨脆性及骨折风险增加。2018年国家卫生健康委发布的最新流行病学调查报告显示,我国40~49 岁人群中OP 的患病率为3.2%,50 岁以上人群为19.2%,65岁以上则高达32.0%[1],随着年龄的增长OP 患病率逐级递增,提示衰老与OP的发生密切相关。
骨髓间充质干细胞(bone marrow mesenchymal stem cells, BMSCs)是一种具有多向分化潜能的细胞,其成骨分化是关系机体骨形成、维持骨代谢正常的重要环节。近年来研究发现,BMSCs 在OP 的防治过程中表现出巨大潜力,在各种骨组织工程或骨修复工程中被广泛运用,但其面临由于细胞衰老引发的活力下降或成骨分化能力不足等问题[2]。因此,深度解析BMSCs的衰老机制,以此探寻OP的防治策略,可能是一个重要的突破口。基于此,本文重点关注OP 与BMSCs 衰老的关系及BMSCs 衰老的机制,以期为OP的防治研究提供更多的参考依据。
1 OP与衰老的关系
有研究对比了平均年龄为78 岁的老年健康女性和平均年龄为27 岁的年轻健康女性骨组织中衰老相关指标的表达,发现老年女性骨组织中不仅衰老相关因子p16 和p21 的表达显著增加,还伴有12个衰老相关分泌表型相关因子表达的显著上调。该团队接着在小鼠研究中也发现,通过对6 月龄、24 月龄小鼠的骨细胞、成骨细胞、成骨祖细胞及BMSCs中的衰老相关因子含量进行分析,发现p16 的表达在24 月龄小鼠的上述骨系细胞中均显著增加[3]。Valdes 等[4]开展了2 150 名18~79 岁的女性流行病学相关研究,发现拥有更长的端粒长度的女性其临床患OP 的风险也就越低;患有OP 的女性与正常女性之间的端粒酶长度差异相当于5 年端粒的衰老程度。由上可知,OP的发生与衰老具有密切关联。
2 BMSCs衰老与OP的关系
BMSCs 是维持骨代谢平衡的关键干细胞,因其拥有较好的干细胞植入特性及多向分化潜能等特点而备受关注。更为重要的是,BMSCs 的成骨分化是成骨细胞祖细胞的重要来源;成骨细胞则是机体合成骨质、调控矿化、促进骨形成的关键功能细胞[5]。因此,足够来源的成骨细胞是保证机体骨骼重建,维持骨代谢平衡的关键。
BMSCs进入衰老状态后,其成骨分化能力明显减弱,既往的研究广泛报道了BMSCs 衰老影响包括OP在内的骨代谢性疾病的进程[6]。研究显示,老年大鼠不仅出现骨代谢紊乱,其来源的BMSCs也出现衰老相关基因p16表达增加、线粒体皱缩等明显衰老特征[7]。同样,OP 大鼠来源的BMSCs 也表现为成骨分化能力显著降低,衰老分泌相关表型明显增加,其线粒体功能也遭到明显破坏[8]。通过DNA 损伤或端粒侵蚀等方式诱导的BMSCs衰老均可导致骨形成能力降低、破骨细胞骨吸收作用增强及OP 的发生[9]。由此可见,BMSCs衰老可能是OP骨代谢失衡的重要病理机制。
3 BMSCs衰老诱发OP骨代谢失衡的相关机制
3.1 自由基学说 生理状态下,生物体内抗氧化酶、非酶抗氧化剂与自由基的合成代谢处于动态平衡;但随着年龄增长或受到某些衰老信号的刺激,使得机体抗氧化剂合成不足或自由基过量产生,引起多种细胞器遭到破坏,尤其是线粒体功能的损伤,引发糖、脂质等物质的合成与代谢紊乱,导致机体引发OP等衰老相关疾病。
BMSCs 的增殖和成骨分化与细胞内的氧化应激水平密切相关。通常,活性氧的增加会抑制BMSCs增殖,使细胞偏向于成脂分化,而成骨分化能力明显减弱,细胞的免疫调节功能也会受到抑制,细胞衰老相关的表型更为显著。有研究对衰老的BMSCs进行细胞周期检测,发现快速老化小鼠来源的BMSCs 细胞周期停滞在G0/G1期,细胞内活性氧增加明显,相关的免疫指标表达也出现紊乱[10]。也有研究显示,清除BMSCs 中过度的活性氧后,BMSCs 的生长及迁移均有明显改善。许多具有抗氧化作用的中药活性成分也被证实通过降低活性氧水平促进BMSCs成骨分化,如白藜芦醇可以通过激活AMPK 信号通路抑制细胞内活性氧的产生,从而逆转BMSCs 衰老[11]。NAD+及NAD+/NADH 的比例是调节自由基氧化重要的介质,在衰老BMSCs 中检测到其NAD+及NAD+/NADH 比例明显升高;对衰老的BMSCs 进行外源性NAD+补充后,其成骨分化可以得到明显改善[12]。
3.2 DNA 损伤应答 DNA 分子损伤的蓄积及修复能力的减退也是影响细胞衰老的重要因素。在细胞受到某些信号刺激后,含有DNA 基因组的DNA 骨架会发生异常,引起双链或者单链DNA 的断裂,并激活机体启动相应的修复机制,引起细胞内的系列反应,这些反应也被统称为DNA 损伤应答。在衰老的细胞中可稳定地观察到DNA 损伤应答病灶中存在断裂双链DNA。利用辐射引起BMSCs 损伤后,发现BMSCs 的增殖能力及基因组DNA 损伤明显,相应的成骨分化能力也受到抑制[13]。组蛋白H2AX 作为DNA 损伤应答的重要组成部分,可以在羧基末端实现快速磷酸化,从而在相应的双链DNA 断裂位点产生γ-H2AX。H2AX 与许多关键的DNA 损伤修复蛋白功能的发挥密切相关,通常H2AX 作用于DNA 损伤修复的早期阶段,在衰老BMSCs中检测到γ-H2AX表达明显增高[9]。
BMSCs 通过持续的DNA 损伤反应激活驱动过早衰老,其特征为DNA 合成抑制,衰老因子p21 和p16蛋白表达及β-半乳糖苷酶活性增加,细胞成骨分化能力降低[14]。传代次数少的BMSCs相比传代次数多的BMSCs 更能抵抗辐射或基因毒性药物引起的DNA 损伤,且γ-H2AX 在传代次数少的BMSCs 中含量更高,在传代次数更多的BMSCs 中含量明显降低[15]。另有研究显示,小鼠BMSCs 在长期扩增培养中逐渐丧失识别内源性和辐射诱导的DNA 双链断裂的能力,这种受损的DNA损伤反应表现为γ-H2AX修复数量的减少,这与DNA 修复动力学的减慢以及DNA双链断裂数量增加有关[16]。
3.3 长寿基因与衰老基因 在细胞衰老过程中,研究者发现沉默信息调节因子(silent information regulator, SIRT)及系列衰老基因(如Klotho、p16、p21、p53等)与机体衰老有密切关联,在BMSCs的衰老过程中也扮演着重要角色。
3.3.1 SIRT家族 哺乳动物的sirtuins家族共包含7个成员,多位研究者已论证过其中的SIRT1、SIRT3和SIRT6 参与细胞衰老的调控。其中,SIRT1 主要通过调控细胞代谢、抑制炎症和细胞凋亡来改善衰老进程[17];SIRT3主要通过调控细胞代谢及氧化平衡发挥抗衰老作用[18];SIRT6通常作用于应激胁迫或代谢相关的基因,加速DNA及碱基的损伤修复,以此来增加染色体稳定性[19]。也有研究显示SIRT2 可以通过赖氨酸去乙酰化改变蛋白活性调控衰老,但现有文献只报道了SIRT2 可能通过抑制破骨细胞活性缓解增龄性OP[20],其与BMSCs的调控关系暂未见相关报道。
研究发现,全身性敲除、成骨细胞或破骨细胞特异性敲除SIRT1基因小鼠较野生型小鼠骨量均明显降低[21];在骨形态发生蛋白9(bone morphogenetic protein-9, BMP-9)诱导的BMSCs 成骨分化过程中,SIRT1 表达明显降低;SIRT1 过表达后,BMSCs 的成骨分化能力显著提高,极大地缓解了25 羟基维生素D-1α 羟化酶敲除小鼠的骨量丢失[22]。SIRT1 还可通过作用于成骨转录因子Runx2 及Sp7 上的乙酰化组蛋白H3K14ac,使得BMSCs的成骨分化能力增强[23]。
SIRT3 含量与细胞中活性氧减少和DNA 损伤加重有关,与BMSCs 衰老程度呈负相关;对BMSCs 补充外源性SIRT3,可明显改善自然或过早衰老BMSCs的衰老相关表型[24]。此外,SIRT3的补充还可通过提高超氧化物歧化酶2 表达以降低细胞内活性氧水平,从而维持细胞内氧化和抗氧化平衡,减轻BMSCs的氧化应激损伤[25]。SIRT3 也可通过激活锰超氧化物歧化酶和过氧化氢酶的产生,促进BMSCs增殖[26]。
随着年龄的增长,BMSCs 中SIRT6 的含量逐渐降低,细胞的迁移及增殖能力也随之降低。条件性敲除BMSCs 中SIRT6的小鼠,不仅其原代BMSCs 表现为成骨分化能力明显降低,还会出现严重的OP 样特征[27]。沉默BMSCs 中的SIRT6后也发现细胞的增殖、迁移和氧化应激耐受能力均显著下降,β-半乳糖苷酶活性提高,衰老因子p16等表达显著增加[28]。
3.3.2 衰老因子 Klotho 是一种与衰老相关的因子,在骨中具有一定的表达丰度,Klotho 能有效抵抗氧化应激等引起的病理状态[29]。有研究显示,加入外源性Klotho 刺激后,BMSCs 的成骨分化及成纤维细胞生长因子受体1/细胞外信号调节激酶信号通路相关因子均受到明显抑制;而沉默BMSCs 的Klotho基因后,BMSCs的成骨分化则明显增强[30]。
在衰老的BMSCs 中,衰老相关因子p16、p21 和p53 的表达也会显著增加。p16 作为细胞衰老调控的重要成员之一,可与细胞周期蛋白D(cyclin D)协同调控细胞周期蛋白依赖性激酶4(cyclin-dependent kinase 4, CDK4)的表达,对细胞的增殖发挥调控作用。对全身性敲除p16基因的小鼠进行双侧去卵巢手术后,发现其超氧化物歧化酶1和2的蛋白表达水平明显上调,而活性氧水平、p21 含量及骨组织中β-半乳糖苷酶阳性骨细胞占比均明显降低[31]。p21 则可抑制与细胞周期相关的cyclin-CDK 复合物,OP 患者骨组织p21 表达量较正常志愿者明显增加[32];另有研究显示,通过靶向清除p21 阳性衰老的BMSCs可减缓辐射诱导的OP和骨髓脂肪增加[33]。p53参与细胞分化、凋亡及DNA 修复等过程,其介导的多条信号途径在细胞衰老过程中发挥着重要调控作用。在全身性敲除p16基因小鼠中cyclin D、p53 及CDK的表达均明显增加[34]。
3.4 端粒与端粒酶 端粒是一段DNA-蛋白质复合体,位于真核细胞染色体末端,由重复的六聚体核苷酸序列(TTAGGG)和相关保护蛋白构成。端粒可覆盖和保护真核生物染色体的末端,主要发挥防止染色体融合和降解的作用[35]。在细胞在分裂的过程中,端粒的长度受到细胞分裂的影响,随分裂次数的增多而变短,并到达某个长度后细胞停止分裂。
端粒酶在细胞中则主要发挥端粒延长的作用,端粒酶生物学障碍的BMSCs的细胞增殖能力明显降低,且细胞更倾向于向脂肪细胞和纤维细胞方向分化[36]。这可能与端粒通过促进线粒体损伤引起能量代谢异常,从而加速BMSCs衰老有关[37]。此外,端粒酶复合物的某些基因突变也会引起OP 等骨代谢疾病的发生[38]。这些研究均表明端粒酶功能紊乱可能是调控衰老BMSCs引起OP的原因。
3.5 表观遗传学 表观遗传学是指DNA 序列保持不变,但生物体受到某些信号影响导致基因发生可遗传改变,该过程在机体发育、生长、衰老等过程中广泛存在[39]。目前,DNA 甲基化、组蛋白修饰及非编码RNA是表观遗传学改变的几种主要形式。
3.5.1 DNA 甲基化 DNA 甲基化是指基因的功能或活性出现可遗传变化,它不改变生物体本身的DNA序列,但却可通过不同机制实现对基因的表达调控[40]。有研究发现,在绝经后OP 妇女的骨组织中存在SOST 基因的表观遗传调控机制。通过将36 名健康受试者与27名OP 患者对比,发现OP 患者的SOST(sclerostin)基因启动子区域CpG 甲基化水平增加。此外,研究人员还观察到,OP 患者SOST甲基化水平的升高与骨和血清中SOST 的降低在功能上密切相关[41]。未经诱导分化的BMSCs 相比,成骨诱导的BMSCs 多出了2 319 个甲基化位点,进一步对这些位点进行KEGG分析后,发现富集在分子黏附和Wnt/βcatenin经典信号通路等16个信号通路[42]。还有研究发现DNA m6A 去甲基化酶参与调控BMSCs 的命运和骨脂分化平衡,可能作为OP治疗的潜在靶点[43]。
3.5.2 组蛋白修饰 组蛋白修饰是一种重要的调控染色质结构的途径,对于基因活性的调节具有重要意义。目前,组蛋白修饰主要包括乙酰化和去乙酰化两种形式。在骨重建过程中,组蛋白修饰以不同形式参与其中[44]。
研究发现,组蛋白甲基转移酶Ash1l在骨质疏松小鼠的骨组织中显著降低,对Ash1l 进行敲除后,发现可导致小鼠患有关节炎或遭受严重的软骨和骨骼破坏[45]。表观遗传修饰蛋白赖氨酸特异性去甲基化酶5A[lysine (K) demethylase 5A, KDM5A]的表达在OP 大鼠的骨组织中明显上调,在BMSCs 中过表达KDM5A 可明显抑制BMP-2 诱导的成骨分化;特异性敲除KDM5A后,BMSCs 的成骨分化能力则显著提高;其具体的机制可能是KDM5A 通过降低Runx2启动子上的H3K4me3 水平,从而实现成骨分化的内源性调节[46]。有研究者在Runx2启动子区发现了高丰度的组蛋白赖氨酸表观修饰标志分子,进一步研究发现KDM5B 在骨髓间充质细胞向成骨细胞分化过程中对Runx2基因的表达起着关键的开关作用[47]。
3.5.3 miRNA 表达 miRNA 功能的实现通常是通过调控靶基因mRNA 的转录或翻译水平,从而实现调控作用。研究发现,在衰老过程中,间充质干细胞和其他肌肉骨骼谱系细胞中部分miRNA 的表达发生显著变化,并且这些miRNA 还可靶向调节衰老中固有激活的细胞周期阻滞通路组件[48-49]。研究发现,来自老年小鼠的BMSCs分泌的外泌体中miR-29b-3p的含量显著增加,且长寿基因SIRT1是外泌体miR-29b-3p 的下游靶点,miR-29b-3p 可调节衰老相关因子从而影响BMSCs 衰老[50]。C57BL/6 小鼠骨骼肌和血清来源外泌体中miR-34a-5p 含量随年龄的增长显著增加,这些外泌体通过递送miR-34a-5p 至BMSCs从而降低其降低活力,加速细胞衰老[51]。也有研究观察到老年大鼠来源的BMSCs中miR-31a-5p表达水平显著升高,且BMSCs表现出成脂分化能力增强和衰老指标增加,成骨分化和干细胞特性降低[52]。还有部分miRNA 也可通过直接或间接调节p53和p21通路,调控BMSCs衰老,进而实现对骨代谢的调控作用[53]。
BMSCs 衰老诱发OP 骨代谢失衡相关机制的总结见图1。
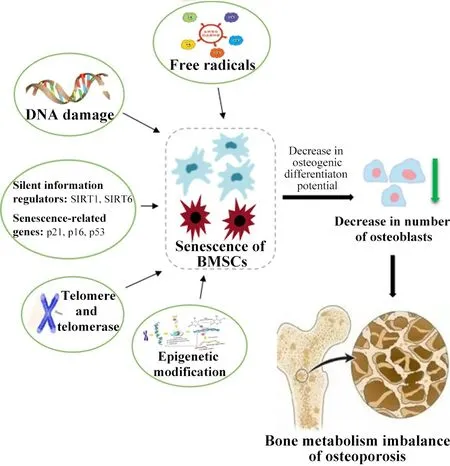
Figure 1.The relationship between the senescence of bone marrow mesenchymal stem cells and the bone metabolism imbalance of osteoporosis.图1 骨髓间充质干细胞衰老与骨质疏松症骨代谢失衡的关系
4 总结与展望
综上所述,BMSCs 是介导机体骨形成、参与骨代谢稳态调控的关键细胞,BMSCs衰老引发的细胞活力降低可能是引发OP骨代谢失衡的重要环节。BMSCs相关衰老机制涉及活性氧、端粒酶、长寿基因、衰老因子等多种因素调控,各种机制既可独立作用,也存在一定关联。现有的研究多是单纯探讨了各机制如何介导BMSCs 衰老,但何种衰老机制在其中占据主导作用及各个机制间如何网络关联暂不十分清楚。因此,探寻衰老的主导作用机制或进一步阐释各衰老机制之间如何相互关联是未来需要开展的重点工作。此外,目前多种中药或中药活性成分表现出了强大的抗氧化、抗衰老作用,且围绕BMSCs、中药活性成分与新型材料已开展了许多研究,并取得了一定的进展。因此,未来的工作也可从该角度深入,开发新型的抗衰老BMSCs 组合材料,为越来越多的BMSCs防治OP等骨代谢性疾病提供新的思路。
猜你喜欢
口腔医学(2021年10期)2021-12-02
癌变·畸变·突变(2019年3期)2019-06-03
中华老年口腔医学杂志(2016年2期)2017-01-15
健康管理(2016年2期)2016-05-30
恋爱婚姻家庭·养生版(2016年5期)2016-05-06
分析测试学报(2015年8期)2016-01-13
中国病理生理杂志(2015年8期)2015-12-21
山东医药(2015年38期)2015-12-07
天津护理(2015年4期)2015-11-10
食品工业科技(2014年13期)2014-03-11
