
1例婴儿期Mowat-Wilson综合征病例报道
2023-10-26 02:39:32李弋飞李春雨姜慧轶
中国实验诊断学 2023年10期
安 阳,李弋飞,李春雨,王 鹤,姜慧轶
(吉林大学白求恩第一医院乐群院区 儿科,吉林 长春130031)
Mowat-Wilson综合征(MWS)为常染色体显性遗传,通常临床表现为特殊面貌,智力障碍,合并其他并发症,包括常见的巨结肠疾病和先天性心脏缺陷或胼胝体发育不全,泌尿生殖系统异常等。现对2022 年11 月吉林大学第一医院收治的1例Mowat-Wilson综合征患儿的临床特点及实验室结果,结合文献进行报道,以提高儿科医师对该病的认识。
1 临床资料
1.1 一般资料患儿,女,6个月,因“间断发热7 d,腹泻4 d”入院。患儿为G1P1,家长自诉产检时曾有“三尖瓣返流”及“侧脑室增宽”,出生体重3.05 kg,生后无窒息史。住院时患儿可抬头,不能自主翻身,不能独坐,可逗笑,能注视、追视,可发出“a、o、ma”等声音。查体:身高68 cm,体重8 kg,一般状态可。特殊面容(前额较突出,眉毛宽,双眼凹陷,眼距宽、内眦赘皮,鼻梁低,鼻小柱突出、右侧外耳廓上缘发育欠佳,下颌呈三角形),头围43 cm。颈部未触及肿大淋巴结,咽部充血。双肺呼吸音粗,无干湿啰音。心率120次/分,律齐,心音有力,胸骨左缘第2肋间可闻及3/6级双期连续性机器样隆隆样杂音。腹软,肝肋缘下2.5 cm,质地中等,脾脏肋下未触及肿大。神经系统查体:颈部无抵抗,克氏征、布氏征阴性,双侧巴氏征阳性,四肢肌力正常,上肢肌张力正常,下肢肌张力欠佳。
1.2 影像学检查腹部及泌尿系彩超未见异常,肺部CT示双肺炎症,心脏彩超示先天性心脏病动脉导管未闭,二尖瓣口、三尖瓣口可见少量返流。头部核磁示脑室系统扩张,双侧侧脑室后角钝圆(图1)。
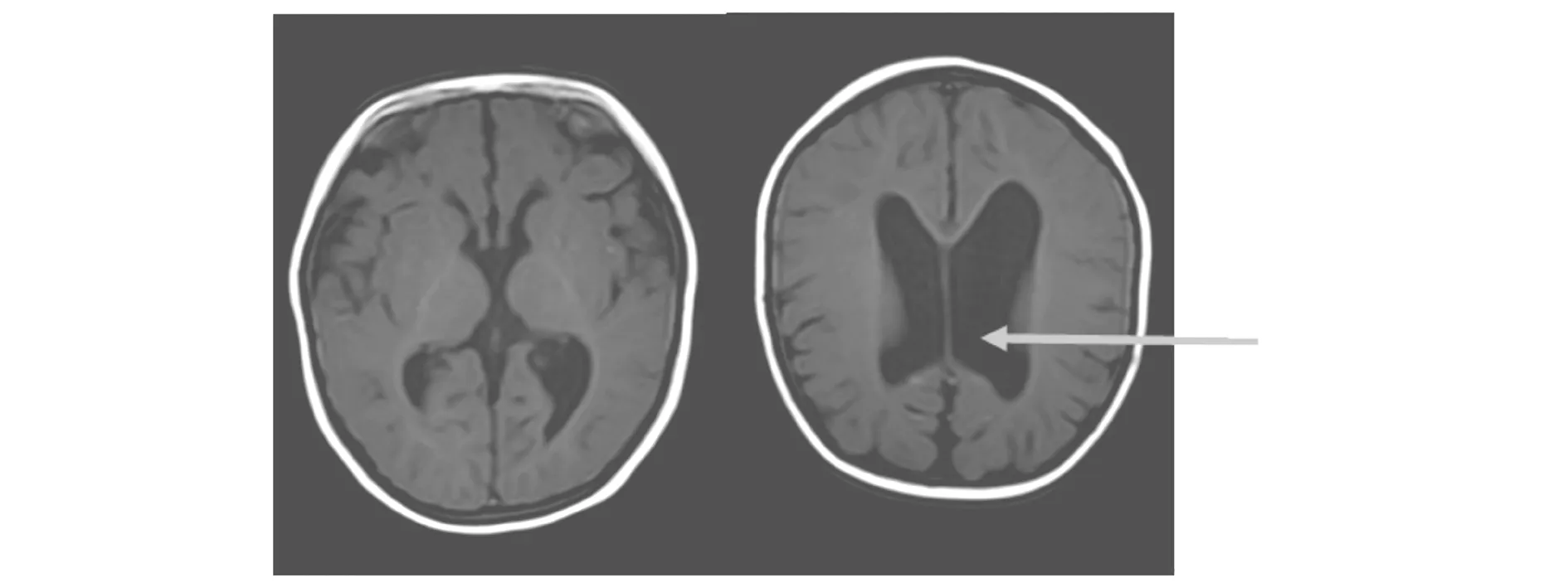
图1 患儿头部磁共振平扫图像(脑室系统扩大,双侧侧脑室后角钝圆)
1.3 实验室检查有机酸及酰基肉碱分析无异常,染色体核型46,XY,染色体未见异常。
1.4 基因检测对患儿及双亲抽取外周血进行全外显子基因组测序检查,检测报告:本次在送检样本中检出1个与受检者提供的临床信息部分相关的致病性变异,基因ZEB2,染色体位于chr2:145147528_145147555,变异信息NM_014795.4C3108_3135delinsTTTGTGTTCGTG(p.Phe1036Leufs*39)杂合,新发变异(图2)。
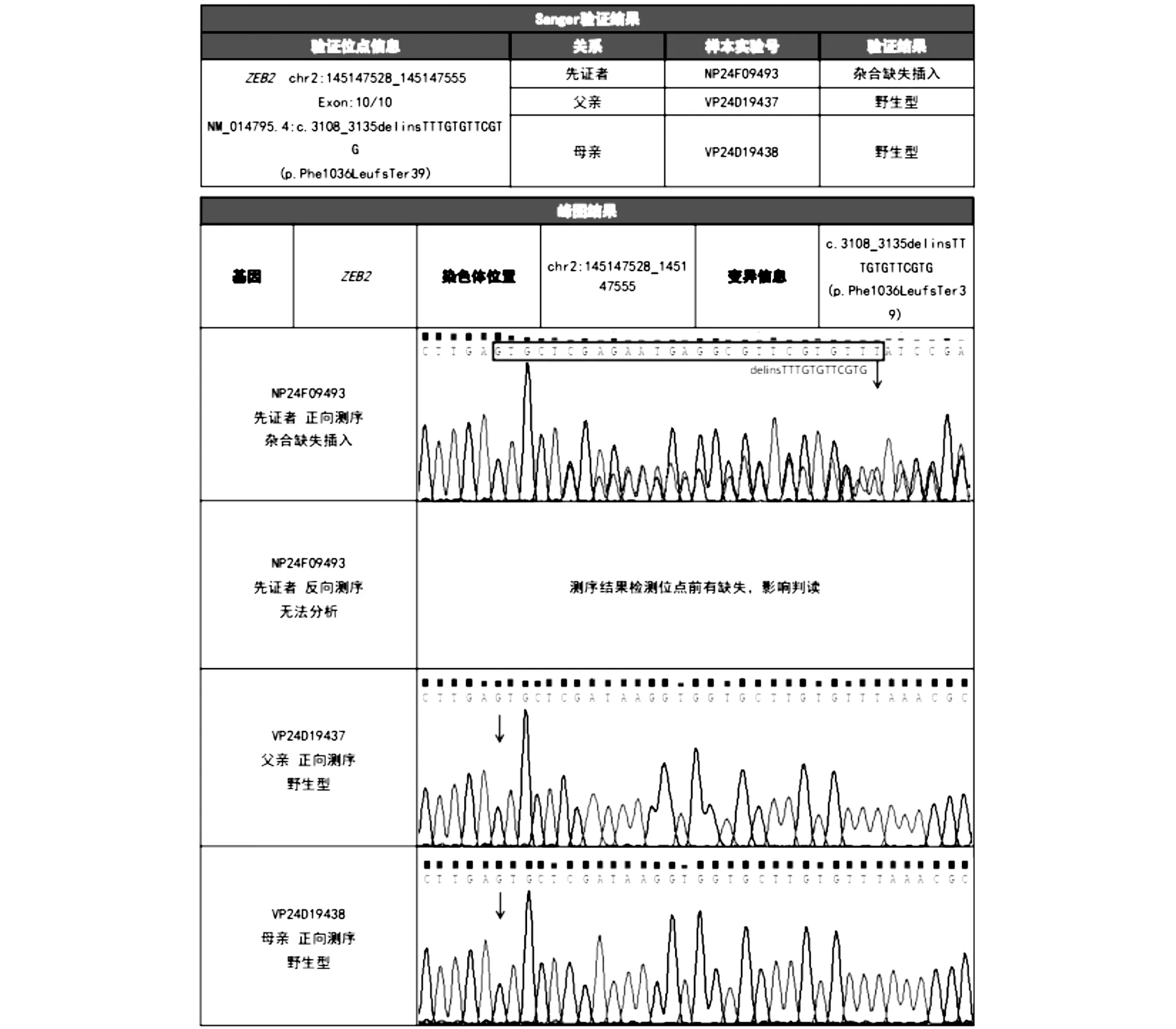
图2 患儿及双亲分子遗传检测报告(患者携带的c.3108_3135delinsTTTGTGTTCGTG 是ZEB2 基因编码区因非三倍数碱基缺失插入导致的移码变异)
2 讨论
Mowat-Wilson 综合征是由 MOWAT等[1]在1998年首次报道的一种常染色体显性遗传病,在西方国家其发病率为 1/50 000~1/70 000[2]。MOWAT等[1]率先描述了6例与先天性巨结肠疾病相关的智力发育迟缓的患者的面部特征,并提出该综合征可能是由染色体2q21-2q23的微缺失或该区域内的一个基因的新生突变引起的。2001年,WAKA等[3-4]发现导致该综合征的原因是ZFHX1B基因(也称为ZEB2基因或SIP1即Smad 交互蛋白1)的缺失或基因内突变。截至目前,已确定约350例MWS患者[5-7]存在ZEB2蛋白编码序列突变。包括全基因缺失(19%)、无义突变(34%)、小插入或缺失导致基因移码突变(40%),后者突变会过早终止密码子,导致蛋白质截断,使其功能丧失[8]。ZEB2(zinc finger E box-binding homeobox 2)定位于 2q22.3,ZEB2有10个外显子,起始密码子位于第2外显子,终止密码子位于第10外显子,ZEB2基因全长约70 kb,编码由1214个氨基酸组成的AEB2蛋白[9]。ZEB2是锌指结构转录因子家族的一员,ZEB2蛋白与SMAD蛋白相互作用,并作为转录抑制因子影响TGFβ信号通路[8-9]。SMAD蛋白是受到严格控制的细胞质介质,在将 TGF-β信号从细胞表面受体传递到细胞核中发挥重要作用[10]。同时ZEB2在正常的胚胎发育过程中至关重要,ZEB家族成员诱导上皮细胞向间充质转化(EMT),这是一个将上皮细胞通过特定程序转化为间充质细胞的过程,对原肠胚、神经嵴、心脏形态、肌肉骨骼系统的形成和颅面结构等发育过程至关重要[11-14]。此外,在人类胚胎中发现ZEB2从中脑到脊髓的中枢神经系统(CNS)中均匀表达。ZEB2已被证实在许多CNS部分的发育中起作用,是神经系统发育过程中的多功能调节因子[15],如调节皮质-皮质下连接、髓鞘形成和海马的发育[16-18]。
MWS是一种罕见的遗传性疾病,目前还没有建立明确的诊断标准,先天性巨结肠病最初被认为是该综合征的一个必要特征,因此命名为严重智力发育迟缓-巨结肠疾病综合征。后来,临床医生发现诊断并不一定基于巨结肠[6,19],其诊断是基于特征性的面部改变伴智力发育迟滞合并其他并发症,包括常见的巨结肠疾病、先天性心脏缺陷或胼胝体发育不全、泌尿生殖系统异常等,最终结合ZEB2的基因改变而确诊该疾病,是典型的基因协助诊断疾病[20]。因此建议对患有某些并发症伴特定面容、智力发育迟滞的患者应进行遗传学分析。迄今为止,国外已报道了超过300名患者患有MWS[21]。我国的MWS发病率低于白种人,截至目前,中文文献只有32个MWS个体被报道[22-33]。其主要原因可能是MWS的正式临床诊断标准尚未建立,临床医生缺乏对该疾病的了解,且大多数情况下缺乏基因检测。
MWS在所有病例中都报道了独特的面容外观[5-6],圆形或方形的脸、眉毛重、内眦赘皮、耳垂大和耳垂上升、鼻子相对较小、鼻根宽、鼻尖较低、鼻翼突出、小而尖下巴[5,34-35]。可能存在牙齿异常,如牙齿拥挤、牙齿错位、牙齿萌出延迟。在ALEKSANDRA JAKUBIAK等[36]研究中,该文章82%的病例中有小头畸形。
除特定面容外,所有患者均伴有发育迟缓和随后的智力低下[5,19]。MWS患者的语言发育严重受损[5,35],大运动落后,平均行走年龄为3.5岁,甚至部分患者没有行走能力[18,35]。MWS患者的性格通常开朗、善于社交。部分患者具有刻板的运动、重复行为和较高的疼痛耐受性[2,18]。大约50%的患者存在先天性巨结肠病,约30%的病例存在无巨结肠的便秘[19,37]。心脏缺陷在MWS患者中也很常见,约占50%~80%,通常包括肺动脉和瓣膜缺损、房室间隔缺损、动脉导管未闭、法洛四联症和主动脉缩窄[18,35]。肌肉骨骼异常最常见的是纤细的手指、轻度跟骨外翻、长脚趾、扁平足和脊柱侧弯[5]。MWS中,脑结构性异常也很常见,可以在颅内MRI 上检测到,有94%的MWS患者颅内MRI结果异常,在研究的54例MWS患者中,胼胝体异常(79.6%)、海马异常(77.8%)和脑室扩大(68.5%)是最常见的脑结构性异常[38],同时也常可见癫痫发作[39]。患儿一般出生时身高和体重都在正常范围内,此后,患者身材矮小(身高低于平均值2个标准差),有些患者体重也会减少,约30%的患者低于第三百分位[5]。
我国目前发表的32例[22-33]中文文献中,均有特殊面容及智力低下,其他并发症包括先心病(68.8%,22/32)、巨结肠(50%,16/32)、便秘(18.8%6/32)、幽门狭窄等消化系统疾病(9.4%,3/32)、癫痫发作(40.6%,13/32)、斜视(3.1%,1/32)、泌尿生殖系统畸形(18.8%,6/32)、胼胝体改变(40.6%,13/32)、侧脑室扩大(12.5%,4/32)、脑白质改变(9.4%,3/32)、脑外间隙增宽(6.3%,2/32),其比例不同可能跟数目较少及种族差异有关。
本例患儿具有典型面部特征,语言运动发育迟缓、且伴有先天性心脏病,符合MWS 的临床特征。该患者ZEB2基因变异,变异信息NM014795.4C3108_3135delinsTTGTGTTCGTG(p.Phe1036Leufs*39),该变异为缺失插入,受检者携带ZEB2基因编码区因非三倍数碱基缺失插入导致的移码变异,即通过无义介导的mRNA降解或编码氨基酸序列的提前终止,导致正常蛋白功能丧失。经Sanger测序验证,该变异为新发变异。该变异未见文献及大规模人群频率数据库。根据现有证据,该变异被定义为疑似致病变异[40]。此新发突变对于扩大该综合征的分子谱和可能了解更多的基因型-表型相关性有着重要作用。
由于本病属于罕见病,目前治疗主要还是以对症支持及个体化康复训练为主,合并先天性心脏病、巨结肠、生殖泌尿系统异常的MWS患者必要时需手术治疗。
猜你喜欢
天津医科大学学报(2021年4期)2021-08-21 02:14:52
心肺血管病杂志(2020年5期)2021-01-14 00:43:30
中国民间疗法(2020年22期)2021-01-07 07:39:34
小天使·二年级语数英综合(2017年4期)2017-04-18 17:29:21
小天使·四年级语数英综合(2017年4期)2017-04-18 09:15:43
中国当代医药(2015年30期)2015-03-01 02:08:19
中国卫生标准管理(2015年3期)2015-01-27 00:57:55
西南军医(2015年2期)2015-01-22 09:09:46
少年文艺·开心阅读作文(2014年5期)2014-10-08 16:11:31
中国医学科学院学报(2010年6期)2010-03-25 13:58:26
