
分散固相萃取结合UPLC-MS/MS测定不同人参加工品中46 种皂苷类化合物
2023-10-21 03:14:46王艳红吴雨桐曹虹芳许煊炜李月茹
食品科学 2023年18期
王艳红,吴雨桐,曹虹芳,许煊炜,赵 丹,李月茹,*
(1.吉林农业大学 农业农村部参茸产品质量监督检验测试中心,吉林 长春 130118;2.贵州中医药大学药学院,贵州 贵阳 550025;3.吉林农业大学中药材学院,吉林 长春 130118)
人参是五加科植物人参(Panax ginsengC.A.Meyer)的干燥根和根茎,具有大补元气、复脉固脱、补脾益肾、生津养血、安神益智之功效[1],是我国传统的名贵中药材,一直以来素有“中药之首,百草之王”的美誉。现代药物化学和药理学研究结果表明,人参中皂苷类物质是其主要的活性成分,具有抗肿瘤、抗衰老、抗氧化、降血糖和增强免疫功能等多种药理作用[2-3]。人参皂苷根据在人参中的量被分为常量人参皂苷(主要人参皂苷)和稀有人参皂苷,常量人参皂苷如人参皂苷Ra1、Ra2、Ra3、Rb1、Rb2、Rb3、Rc、Rd、Re、Rg1、Rg2、Rf等含有较多糖基、在人参中含量相对较高,但其生物活性较低,在人体内的吸收率也很低;稀有人参皂苷如人参皂苷Rg3、Rg5、Rg6、F4、Rh1、Rh2、Rh3、Rh4、Rk1、Rk2、Rk3、F1、F2等在人参属植物中含量很少,很难从人参属植物中直接分离得到,一般通过降解常量人参皂苷的方法制备稀有人参皂苷,但稀有人参皂苷含糖基较少,生物活性更好,身体吸收率更高[4-5]。此外,人参通过不同的加工方式,部分常量皂苷也会发生转化,产生稀有人参皂苷[6-7]。市场上最常见的人参加工产品为生晒参和红参,近年来,人参经过九蒸九曝加工方法而成的黑参也逐渐出现在市场上,且由于黑参除了部分稀有皂苷含量较高,还含有一些特异性的稀有人参皂苷,并表现出更强的生物活性,越来越受到广大消费者的欢迎[8]。可见不同的加工方式引起了人参皂苷含量和种类的差异,对其药理活性和临床应用也有较大的影响。为此,评估人参皂苷水平对人参加工产品的质量控制和正确有效使用至关重要。而人参皂苷种类繁多,且结构具有多样性和相似性,针对人参相关产品复杂体系中多种皂苷类成分的快速鉴定及定量分析检测技术,国内外研究者们也一直在不断的探索和研究。
近20年来,利用高效液相色谱法和超高效液相色谱法定量分析人参皂苷的含量已经是一种比较成熟的分析手段[9-12],但由于检测种类有限,分析时间较长,单一液相色谱方法难以满足人参及人参相关产品复杂体系中多种皂苷类成分同时快速分析检测。随着现代质谱技术的快速发展,液相色谱-质谱联用技术因其具有灵敏度高、分辨率好且高效快速便捷等优点已被广泛用于人参皂苷定性定量分析研究中。目前,虽然已有大量研究报道了应用液相色谱-质谱联用技术分析及鉴定人参皂苷类成分的检测方法,从液相色谱-质谱联用技术,到超高效液相色谱-高分辨质谱联用技术等。但既简单、准确、快速又覆盖皂苷种类多的定量检测分析方法还有限。三重四极杆质谱通常采用的多反应监测(multiple reaction monitoring,MRM)模式需要已知的标准品优化仪器参数和对分析物进行定量分析检测,具灵敏度高、选择性强的优势,更适用于已知物质的定量分析。如Xia Yonggang等[13]采用超高效液相色谱结合电喷雾电离和三重四极杆质谱分析测定了西洋参中22 种人参皂苷的含量;石婧婧等[14]建立了超快速液相色谱-三重四极杆/线性离子阱质谱法同时测定红参中17 种人参皂苷类成分;赵立春等[15]建立了人参中28 种人参皂苷含量的超高效液相色谱-质谱法(ultra-high performance liquid chromatography-tandem mass spectrometry,UPLC-MS/MS)检测方法。这些超高效液相色谱-三重四极杆质谱法,操作简单、快速准确,但是分析皂苷种类和数量覆盖面较窄,难以满足人参不同产品中皂苷类化合物复杂多样的分析检测要求。郝颖[16]和戴雨霖[17]等采用了高分离度快速液相色谱-四极杆-飞行时间质谱法分析鉴定了不同人参样品中40多种皂苷类成分;Lee等[18]采用超高效液相色谱-高分辨飞行时间质谱联用技术分析鉴定了人参根、茎、叶和果中的58 种皂苷类化合物。这些高分辨质谱法检测人参皂苷的多组分常采用飞行时间质谱检测器,具有超高的分辨率和精确分子质量测定功能,更适用于未知物质的定性鉴别。且超高效液相色谱-高分辨质谱联用仪不仅价格昂贵,还需具备较高的专业技术水平等,在大部分领域的应用仍处于发展阶段,大部分实验室仍未普及,液相色谱-三重四极杆串联质谱仪仍然是当前科研和检测机构的主流定量分析手段。
由于人参不同产品中含有较多的糖类、脂类及色素等成分,为了降低基质的干扰,保证数据的准确性,并减少对色谱柱和仪器的污染,需要对其进行净化处理。目前最常见的人参皂苷前处理净化方法仍是传统的固相萃取法[19-20]、液液萃取法[21-22],这些方法存在过程繁琐、耗时长、成本高、需要大量有毒有机溶剂等问题,且无法满足批量样品中多种皂苷的同时简单、快速、准确检测的需求。分散固相萃取法作为一种简便、快速、价格低廉的分析方法,已成功用于农药残留检测领域。近年来该方法在中药有效成分提取净化及分析中的应用研究也有报道,如药用狗牙花中生物碱、五味子中木脂素[23-24]。但将分散固相萃取法用于人参中皂苷类成分净化分析方面的研究鲜见报道。本研究在前人研究基础上,采用了分散固相萃取法对人参样品进行净化处理,利用超高效液相色谱-三重四极杆串联质谱仪建立46 种皂苷类成分的检测方法,并对人参不同加工品中皂苷类化合物的含量和组成进行检测分析和比较。该方法进一步拓展了现有方法的检测范围,增加了样品前处理的净化技术和皂苷类化合物的检测种类,操作简便、快速,为不同人参加工品中多种皂苷类化合物的快速分析检测提供了技术参考。
1 材料与方法
1.1 材料与试剂
3 种人参加工品(生晒参、红参和黑参)为吉林省抚松万良人参市场随机购买。
46 种皂苷标准物质对照品(纯度均不小于95%)购于上海源叶生物科技有限公司和成都曼斯特生物科技有限公司。
甲醇、乙腈(均为色谱纯) 德国Merck公司;甲酸(色谱纯) 美国Sigma-Aldrich公司;其他试剂均为分析纯。
N-丙基乙二胺(primary secondary amine,PSA)、十八烷基键合硅胶(C18)吸附剂、石墨化碳黑(graphitized carbon black,GCB)吸附剂 北京恩加壹科技有限公司;实验用水为超纯水(Milli-Q纯水处理系统制备,美国Millipore公司)。
1.2 仪器与设备
Sep-pak C18固相萃取小柱(1 g/6 mL) 美国Waters公司;LC-30A超高效液相色谱仪 日本Shimadzu公司;AB QTRAP 4500三重四极杆/复合线性离子阱质谱仪、Analyst、Peak View和MultiQuant数据处理系统 美国AB SCIEX公司;Kinetex XB-C18色谱柱(2.1 mm ×100 mm,2.6 µm) 美国Phenomenex公司;KQ-500DE数控超声波清洗器 昆山市超声仪器有限公司;Heraeus Multifuge XIR台式冷冻离心机 美国Thermo公司;AL204型电子天平 美国Sartorius公司;VORTEX-6旋涡混合器 北京海天友诚科技有限公司。
1.3 方法
1.3.1 标准溶液的配制
准确称取4 6 种皂苷标准品各1 0 m g(精确至0.1 mg),分别以甲醇溶解并定容,配制成1 mg/mL的标准品储备液,于4 ℃避光保存备用。取适量标准储备液,以甲醇稀释成100 µg/mL的标准中间液。分别取适量标准中间液,根据各化合物的响应情况,分别用甲醇稀释成不同质量浓度的系列混合标准溶液。
1.3.2 样品前处理
提取:取干燥至恒质量的人参加工品样品粉碎,过60 目筛,准确称取1 g(精确到0.000 1 g)样品粉末,加入50 mL 80%甲醇溶液,浸泡过夜,超声40 min,以5 000 r/min离心15 min,精密移取上清液2 mL,60 ℃下氮气吹干,用甲醇溶解并定容至5 mL,待净化。
净化:准确移取提取液2 mL至装有100 mg PSA的离心管中,涡旋振荡2 min,5 000 r/min离心6 min。上清液过0.22 µm滤膜,备用。或根据实际浓度用甲醇适当稀释至线性范围内,作为供试品溶液待测。
1.3.3 色谱条件
Kinetex XB-C18色谱柱(2.1 mm×100 mm,2.6 µm);柱温40 ℃;流速0.3 mL/min;进样量2.0 µL;流动相A为0.1%甲酸溶液,B为乙腈;梯度洗脱:0~1.0 min,81% A、19% B;1.0~2.0 min,81%~72% A、19%~28% B;2.0~14.0 min,72%~65% A、28%~35% B;14.0~20.0 min,65%~55% A、35%~45% B;20.0~25.0 min,55%~43% A、45%~57% B;25.0~28.0 min,43%~0% A、57%~100% B;28.0~30.0 min,0% A、100% B;30.0~32.0 min,0%~81% A、100%~19% B;32.0~35.0 min,81% A、19% B。
1.3.4 质谱条件
电喷雾离子源负离子模式;MRM模式;雾化电压-4 500 V;离子化温度450 ℃;雾化气压力45 psi;辅助气压力4 5 p s i;碰撞气压力M e d i u m;气帘气压力35 psi。
1.3.5 基质效应计算
基质效应(matrix effect,ME)/%=A/B×100。其中A为样品中皂苷类物质的峰面积;B为纯溶剂中同浓度皂苷类物质对应的峰面积。当ME>1时,表示基质增强效应,当ME<1时,表示基质抑制效应。若ME为80%~120%时,表示ME不明显,可以忽略;若ME在50%~80%或120%~150%时,表现为中等ME;若ME<50%或者ME>150%时,则表现为强ME[25]。
1.4 数据处理
通过与仪器配套的AB SCIEX公司的Analyst 1.6软件系统完成数据采集与处理,在Multi Quant 3.0和Peak View 2.0软件上定性定量处理和谱图分析,采用Excel和分析软件SIMCA 14.1对结果进行统计分析及图表绘制。
2 结果与分析
2.1 色谱条件的优化
本实验所测的皂苷类化合物种类多,但结构相似,性质差异较小,且含有多对同分异构体,这些化合物具有相同的前体和产物离子,在质谱上无法通过离子对其进行区分,若要准确定量这些化合物,色谱峰要有一定程度的分离。因此实验对色谱条件进行优化,以实现皂苷类化合物的最佳色谱分离以及各化合物的良好峰形和最佳响应。实验首先考察了在相同参数条件下,不同型号色谱柱Waters HSS T3(100 mm×2.1 mm,1.7 µm)、Waters BEH C18(100 mm×2.1 mm,1.7 µm)和Kinetex XB-C18(2.1 mm×100 mm,2.6 µm)的分离效果,结果表明,HSS T3色谱柱对于部分皂苷类物质分离效果较差,特别是无法实现同时对多对同分异构体化合物的基线分离;而Waters BEH C18和Kinetex XB-C18色谱柱对46 种目标化合物均呈现较好的色谱峰形和分离度,但考虑到对系统压力的影响,选择具有低柱压的Kinetex XB-C18作为分离色谱柱。在流动相选择方面,考察反相色谱常用的有机相溶剂(甲醇、乙腈)与水组成的流动相体系对皂苷类成分的分离效果,发现以乙腈体系为流动相时,系统平衡时间相对较快,且洗脱能力和分离效果均优于甲醇体系。在流动相中加入适量的甲酸可以减少色谱峰的拖尾,提高皂苷类化合物的离子化效率,改善色谱峰形,并增强目标化合物的响应信号和稳定性。因此,本实验流动相选择最常用的乙腈-0.1%甲酸溶液系统,在很大程度上减少了溶剂系统带来的背景和基质干扰。最后,通过调节梯度洗脱程序使互为同分异构体的皂苷类化合物进一步分离,由待测目标化合物的结构可知,本研究中46 种皂苷类化合物中同分异构体较多,但可通过保留时间和离子碎片的不同加以区分。例如,人参皂苷Rb2、Rb3和Rc与人参皂苷Rs1和Rs2这两组同分异构体,它们各自产生的检测离子碎片虽然相同,但可通过保留时间的不同得到较好分离;而另一组同分异构体原人参三醇型皂苷Rf和拟人参皂苷F11,二者保留时间较接近,通过调节流动相梯度也无法得到较好分离,但由于二者产生的离子碎片不同则可加以区分。在优化的仪器条件下,各皂苷类化合物灵敏度和和峰形均可满足检测要求,整个梯度运行时间为35 min,46 种皂苷类化合物均得到有效分离,能够达到准确定性定量目的。46 种目标物混合标准溶液的MRM色谱图见图1,46 种化合物信息见表1。
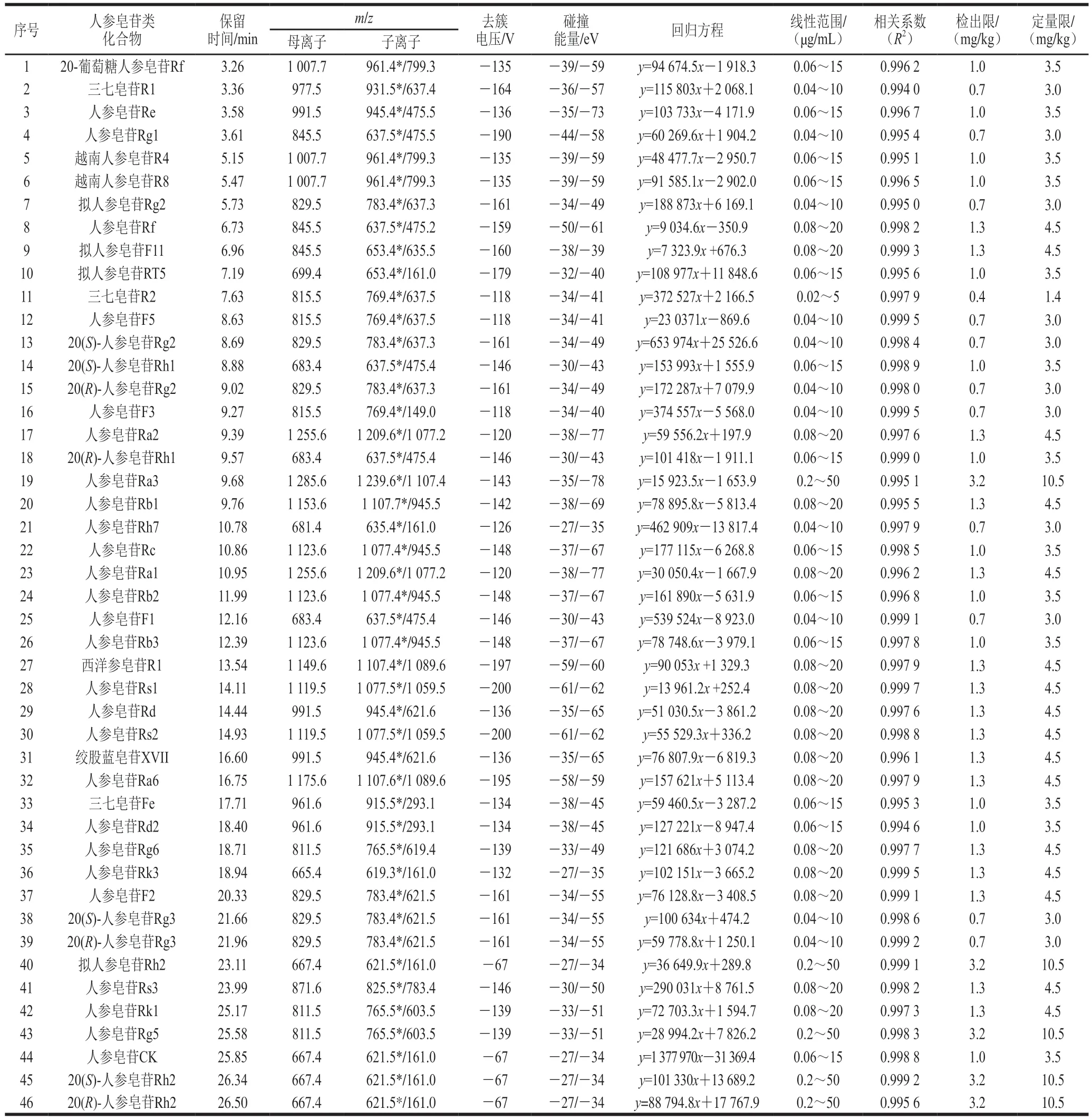
表1 46 种人参皂苷类化合物的质谱参数、线性关系、检出限和定量限Table 1 Mass spectrometric parameters, linearities, LODs and LOQs of 46 ginsenoside compounds

图1 46 种人参皂苷类化合物标准物质的MRM色谱图Fig.1 MRM chromatograms of 46 ginsenoside standards
2.2 质谱条件的优化
采用针泵直接将标准溶液注入离子源的方式对7 种皂苷类化合物质谱参数进行优化。分别将46 种待测皂苷类化合物标准溶液(质量浓度均为1.0 µg/mL)以7 µL/min注入电喷雾离子源,采用正、负2 种离子模式进行扫描,发现皂苷类化合物在负离子模式下具有较高的稳定性和较高的响应值,这与文献[26]相关报道一致,因此选择负离子模式。皂苷类物质在负离子模式下的一级质谱图中,其准分子离子主要以[M-H]-和[M+HCOOH-H]-的形式存在。在Q1进行全扫描中,找出母离子,母离子在Q2打碎后,在Q3进行子离子全扫描以确定各组分的主要碎片离子,选择2 个无干扰、灵敏度相对较高的稳定的特征子离子,组成MRM离子对,作为定性定量的离子对。在MRM模式下优化去簇电压和碰撞能量,最终确定了46 种皂苷类化合物的MRM离子对、去簇电压及碰撞能量。优化后的质谱参数如表1所示。
2.3 提取条件的选择
人参皂苷类化合物的提取相关报道较多,常用的提取溶剂和方法有甲醇回流提取[27]、甲醇超声提取[28]、80%甲醇超声提取[12]、水饱和正丁醇超声提取[29]和60%乙醇超声提取[14]。本研究在前人研究的基础上,选用提取方法较简单的超声提取法,并考察甲醇、80%甲醇、水饱和正丁醇和60%乙醇作为提取溶剂时,各个皂苷类化合物的提取效果。经过对比发现,80%甲醇和60%乙醇对大部分皂苷类化合物的提取效率优于纯甲醇和水饱和正丁醇,但60%乙醇为提取溶剂时,提取液的颜色较浓,杂质较多,且基线不稳定,不利于后续的净化处理,而80%甲醇溶液的提取效果总体最佳,基线和峰形也较好,因此最终选用80%甲醇溶液作为提取溶剂。
2.4 净化条件的优化
人参加工品提取液中的糖类、色素及有机酸等杂质会产生ME,干扰待测物的分析,而且会损坏检测仪器和色谱柱,需要对样品进一步净化。为了使净化时尽可能多地去除杂质干扰,又能减少对目标物的损失,同时使实验方法达到简单、快速、准确,具有时效性。实验对Sep-pak C18(1 g/6 mL)为固相萃取柱的固相萃取法[9]和分散固相萃取2 种净化方式进行比较。发现C18固相萃取柱净化后,各化合物的回收率可达到80%~105%,净化效果也较明显,但C18固相萃取柱净化过程需要活化、平衡、洗脱等多个步骤,处理时间长,过程也较繁琐。分散固相萃取净化方法具有快速、简单、廉价、有效等特点,可将提取液直接达到净化目的,是较为理想的样品前处理净化方法。常用的吸附剂有C18、GCB、PSA等。实验分别考察3 种吸附剂C18、GCB和PSA对46 种皂苷类化合物回收率的影响,结果如图2所示,当使用C18作为吸附剂时,46 种目标化合物的回收率为8.9%~31.6%,这是因为C18吸附剂具有疏水作用,除了能吸附色素、脂肪和维生素等非极性的组分,对中等极性或极性小的分子也有吸附[30],因此对皂苷类化合物的吸附作用较强,不利于净化。当使用GCB作为吸附剂时,净化后的提取液颜色最浅,虽然对人参样品提取液中色素类物质净化效果最好,但由于GCB其表面光滑无孔的石墨晶型结构决定其对非极性化合物、弱极性化合物和部分极性化合物都具有一定的吸附作用[31]。因此GCB不仅对色素类物质吸附性好,还会吸附目标化合物,使目标化合物的回收率降低,GCB对46 种目标化合物净化后的回收率为30.1%~69.5%。PSA对46 种目标化合物的吸附作用均较弱,回收率在80.2%~103.8%之间,而且PSA对脂肪酸、色素、有机酸、及糖等极性干扰物的去除效果较好,基本可满足净化需求[32]。
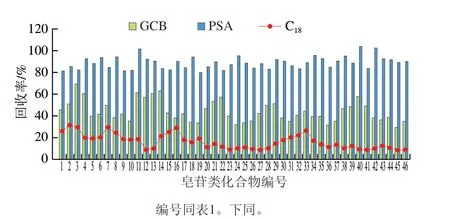
图2 3 种吸附剂对46 种目标化合物回收率的影响Fig.2 Effects of three sorbents on recoveries of 46 analytes
为了进一步提高样品的净化效果,实验对PSA用量进行调整,在2 mL提取液中分别加入60 、80、100、120、140 mg的吸附剂PSA进行净化处理,同样以净化回收率作为考察指标进行比较,结果如图3所示,发现PSA添加量为60、80、100 mg时46 种皂苷类化合物的回收率无明显变化,回收率为81.9%~113.5%,其中100 mg PSA的回收率和净化效果均较好。随着PSA用量的增加,目标化合物的回收率受到严重影响,添加量120 mg PSA的回收率为72.2%~85.8%,添加量140 mg PSA的回收率为65.6%~80.8%,这是由于吸附剂用量过高时,会对目标化合物产生部分吸附,从而降低目标化合物的回收率。因此,综合考虑回收率和净化效果,实验最终选择2 mL提取液中加入100 mg PSA作为净化剂,可以获得良好的净化效果,基质干扰小,且不会对待测皂苷类化合物产生明显吸附。
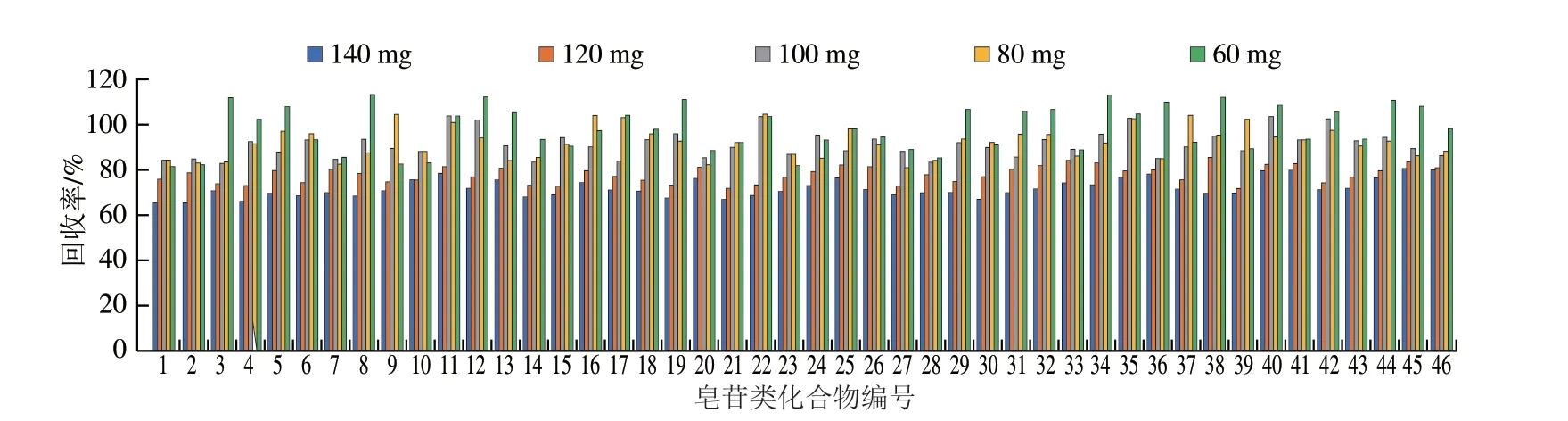
图3 PSA吸附剂用量对46 种目标化合物回收率的影响Fig.3 Effect of PSA dosage on recoveries of 46 analytes
2.5 ME考察
由于人参加工样品基质复杂,可能会对检测结果的准确度造成干扰,为此对方法中的ME进行考察。分别以生晒参、红参、黑参样品为基质,按照1.3.2节样品前处理方法制备基质溶液,分别用基质溶液和纯溶剂配制一系列混合基质标准工作溶液和纯溶剂标准工作液,用于标准曲线的测定。利用基质标准曲线测算出基质中目标物质本底浓度和峰面积,再用纯溶剂标准曲线测算出对应浓度下纯溶剂中目标物质的峰面积,利用公式计算ME。
本研究中分别比较了样品在分散固相萃取法净化前后的ME,如图4所示,分散固相萃取净化前生晒参、红参、黑参样品中部分目标化合物的ME值在120%~150%之间,表现为中等基质增强效应;部分目标化合物的ME值在50%~80%之间,表现为中等基质抑制效应。经分散固相萃取净化后,46 种目标化合物在生晒参、红参、黑参样品中的ME值分别为88.6%~110.9%、86.5%~109.6%和84.8%~112.2%,ME表现不明显,说明样品基质经过前处理过程的分散固相萃取净化和较大体积的稀释,有效降低了人参加工品中ME的影响,保证了定性定量结果的准确、可靠。
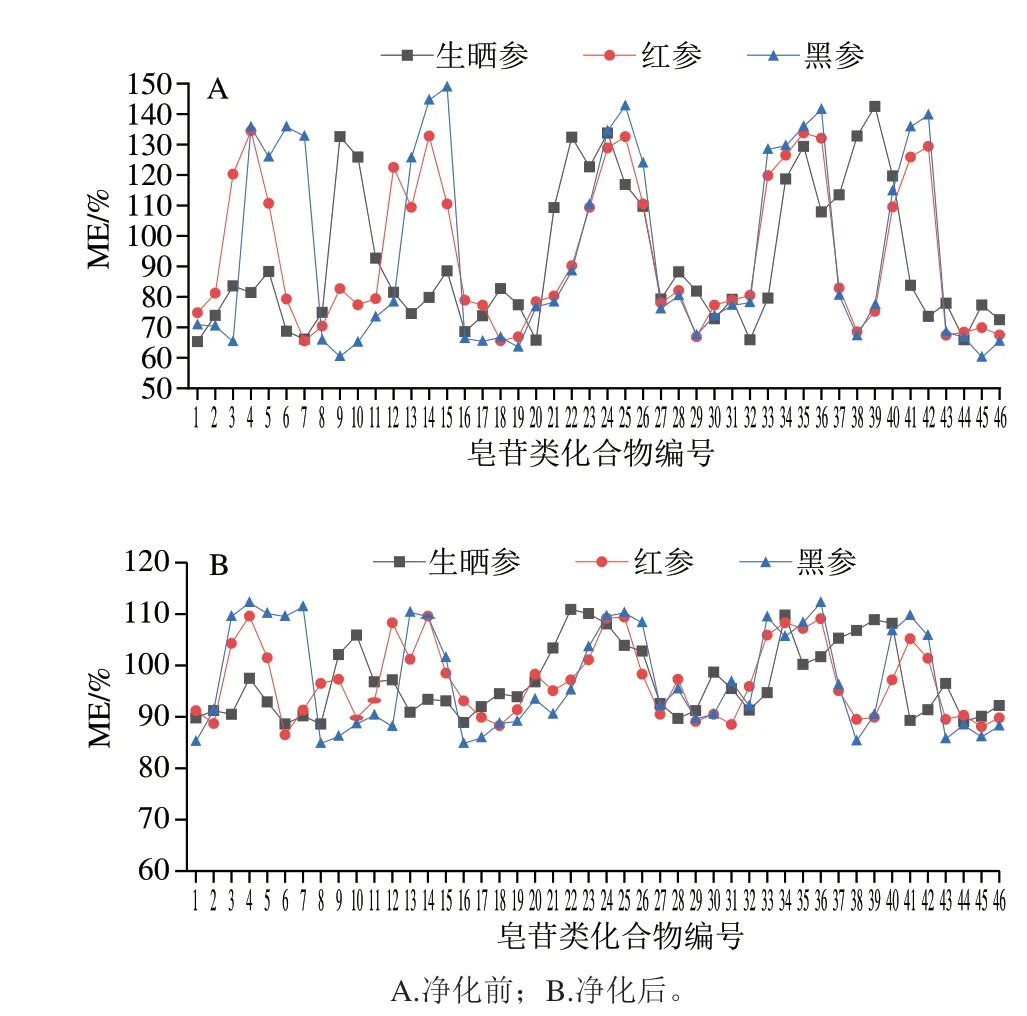
图4 46 种目标化合物MEFig.4 Matrix effects of 46 analytes
2.6 方法学验证
2.6.1 方法的线性范围、检出限及定量限结果
在优化的分析条件下,测定46 种皂苷类化合物系列混合标准工作液,以各目标化合物定量离子对的峰面积为纵坐标(y),以相应的质量浓度为横坐标(x),绘制标准曲线,得到各目标物的线性回归方程。以3 倍信噪比确定方法的检出限,以10 倍信噪比确定方法的定量限。46 种人参皂苷类化合物在0.02~50 µg/mL范围内呈现较好的线性关系,所对应的相关系数(R2)均大于0.99。方法的检出限和定量限分别在0.4~3.2 mg/kg和1.4~10.5 mg/kg范围内,均满足检测需要,结果见表1。
2.6.2 回收率实验结果
在已知皂苷类化合物含量的生晒参、红参和黑参样品中,分别添加低、中、高3 个浓度水平的加标回收实验,每个添加水平重复5 次。计算平均回收率和相对标准偏差(relative standard deviation,RSD),结果见表2。46 种皂苷类化合物除20(S)-人参皂苷Rh2和20(R)-人参皂苷Rh2在3 种人参加工产品中平均加标回收率为80.5%~84.6%,其余44 种皂苷类化合物的平均加标回收率均达到85.2%~111.6%之间,RSD均在1.3%~6.8%之间,表明本实验方法有较好的回收率,结果准确可靠。
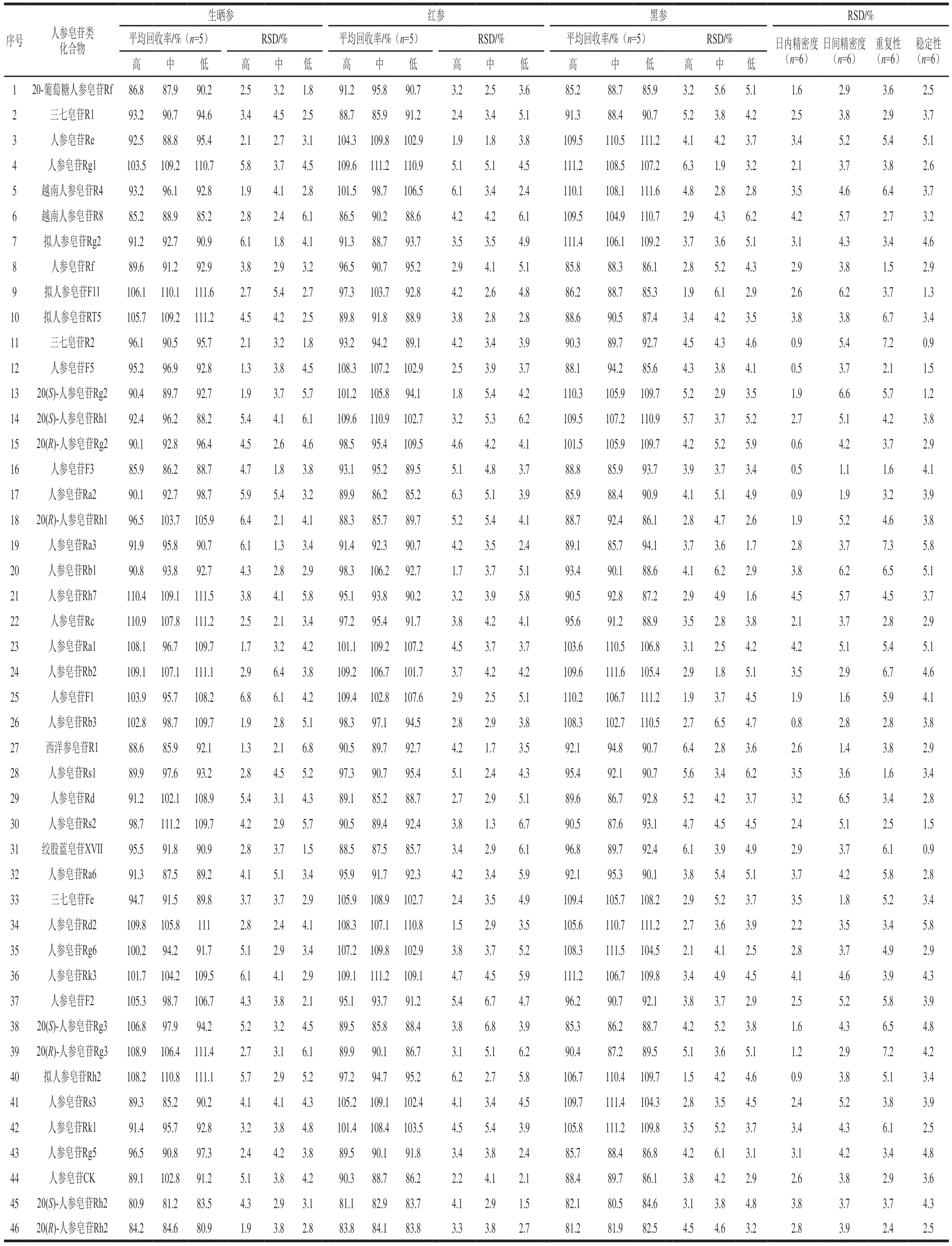
表2 皂苷类化合物的加标回收率、日内和日间精密度、重复性及稳定性Table 2 Spiked recoveries, intra-day and intra-day precision, repeatability and stability for the determination of 46 ginsenosides
2.6.3 精密度、稳定性和重复性结果
取5 µg/mL的混合标准溶液分别在1 d内(6 次)和3 d内重复进样(2 次/d)。计算各成分含量的日内和日间RSD,用以表征日内和日间精密度。取同一样品分析溶液,分别在0、2、4、8、12 h和24 h进样6 次,测定目标化合物峰面积的RSD,考察其稳定性。取同一批样品粉末6 份,按1.3.2节方法制备供试品溶液,在1.3.3节和1.3.4节条件下进样,计算目标化合物测得含量的RSD,考察其重复性。结果见表2。46 种皂苷类化合物的日内精密度和日间精密度分别为0.5%~4.5%、1.1%~6.6%。重复性RSD范围为1.5%~7.3%,稳定性RSD范围为0.9%~5.8%,表明该测定方法具有较高的精密度和较好的稳定性和重复性。
2.7 实际样品分析
应用本方法对市售的15 批生晒参、红参和黑参3 种不同人参加工品进行分析检测,每种人参加工品5 批,每个样品重复测定3 次(图5)。皂苷类成分在3 类人参加工品中呈现明显不同的含量分布特征,结果如表3所示。为了更直观地区分不同加工方式的人参产品,以46 种人参皂苷的含量为变量,采用多元变量统计分析软件SIMCA 14.1对15 份不同人参加工品进行主成分分析(principle component analysis,PCA),见图6。PC1和PC2的累计贡献率达到95.4%,可代表皂苷类成分指标。得分图中生晒参、红参和黑参样品之间相距较远,彼此之间可获得良好区分,表明不同加工方式的人参产品皂苷类成分的含量差异较大。

表3 3 种人参加工产品中人参皂苷类化合物的含量(n=3)Table 3 Contents of ginsenosides in three processed ginseng products (n = 3)mg/kg
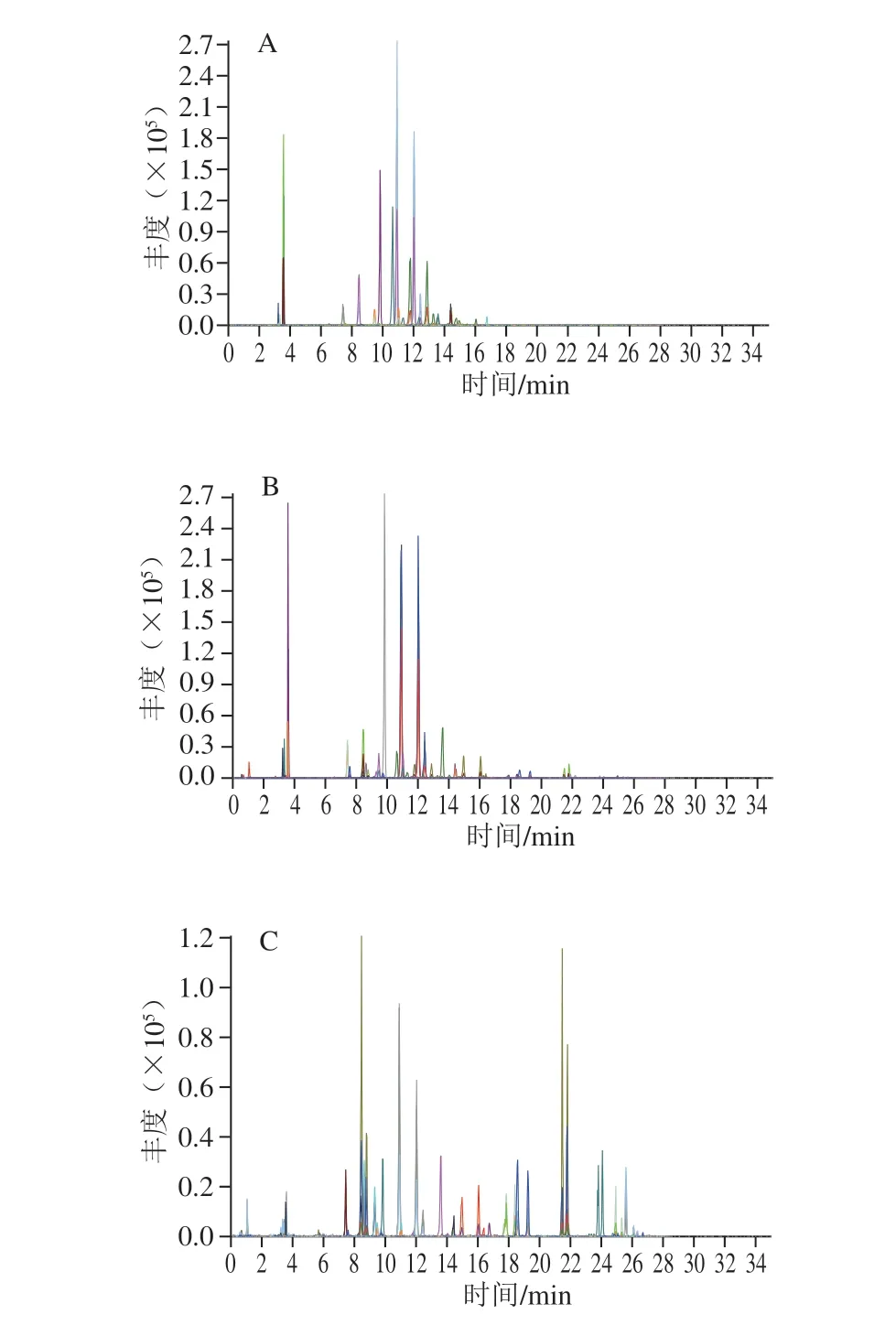
图5 代表性样品的总离子流色谱图Fig.5 Total ion current chromatograms of representative samples
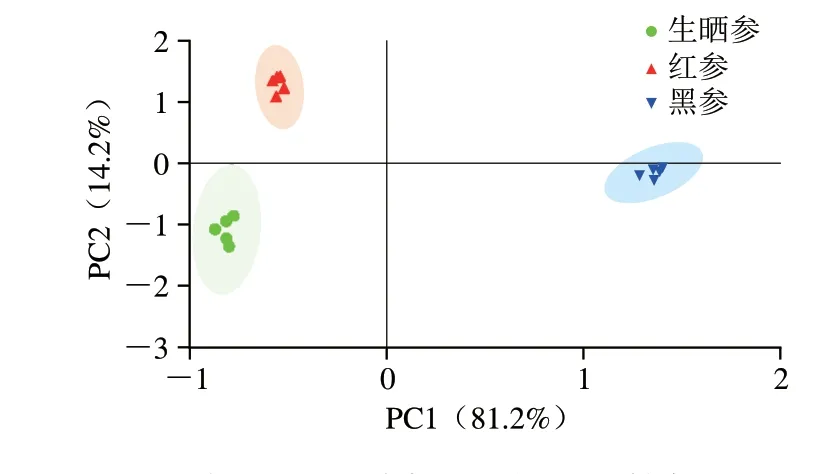
图6 3 种人参加工产品的PCA得分Fig.6 PCA score plot for three processed ginseng products
生晒参是通过在阳光下使鲜人参脱水生产,而红参和黑参是通过不同的高温循环蒸制生产[33-34],这些不同的加工方法诱导了人参皂苷的化学变化,部分人参皂苷转化成了稀有人参皂苷[35]。因此,加工方式的不同是导致人参产品中皂苷类成分含量差异较大的主要原因。
为了进一步明确不同加工方式的人参差异性成分,在PCA基础上,采用变量投影重要性(variable importance in projection,VIP)法筛选,VIP值见图7。以VIP值大于1为标准[36],筛选出造成不同加工方式人参差异的17 个皂苷成分,包括20-葡萄糖人参皂苷Rf、西洋参皂苷R1、人参皂苷Re、Rg1、Rf、Rh1(S)、Ra2、Rb1、Rc、Ra1、Rb2、Rg6、Rk3、Rg3(S)、Rg3(R)、Rk1、Rg5。如图8所示,20-葡萄糖人参皂苷Rf、人参皂苷Re、Rg1、Rf、Ra2、Rb1、Rc和Rb2在生晒参和红参中相对含量较高,在黑参中相对含量较低,这些皂苷均为常量人参皂苷,可作为生晒参和红参的主要成分;而人参皂苷Rh1(S)、Rg6、Rk3、Rg3(S)、Rg3(R)、Rk1、Rg5在黑参中相对含量较高,在红参中相对含量较低,在生晒参中几乎不含,且这些皂苷均为稀有人参皂苷,可见经过蒸制的红参和黑参样品中部分原有的人参皂苷发生了转化,产生了稀有人参皂苷,这些差异性皂苷成分可作为区分黑参、红参和生晒参的标志物群。
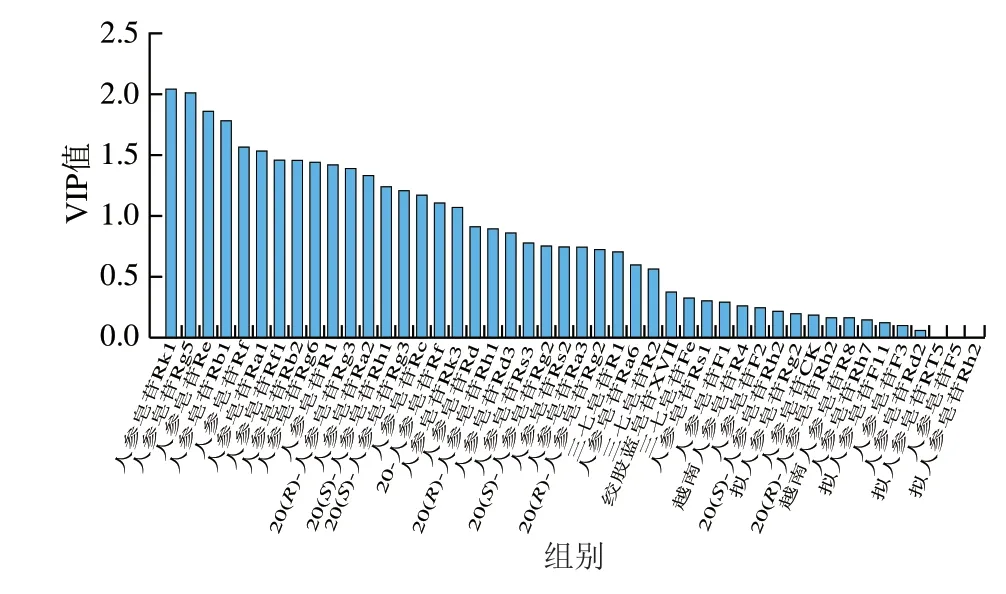
图7 各成分的正交偏最小二乘判别分析VIP值图Fig.7 Orthogonal partial least squares-discriminant analysis VIP plot for 46 ginsenosides
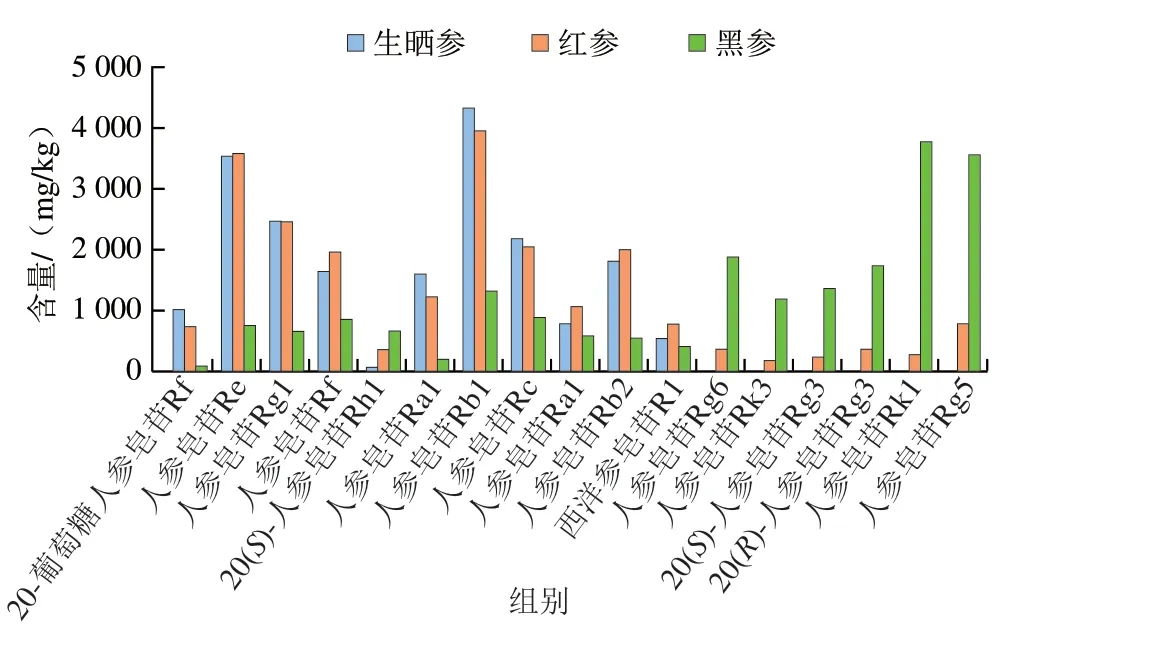
图8 3 种人参加工品中差异性成分含量Fig.8 Contents of differential ginsenosides in three processed ginseng products
3 结 论
本研究采用分散固相萃取净化,建立了人参加工品中46 种皂苷类化合物的UPLC-MS/MS检测方法。实现在35 min内可同时完成46 种皂苷类化合物的快速分析和定量检测,与已有方法相比,本方法引入了分散固相萃取技术,即降低了基质干扰,又能减少对目标物的损失,且简化了操作步骤,使样品前处理更加简便高效,实用性强。通过仪器条件的优化,采用MRM检测,在很大程度上提高了皂苷类化合物的检测通量,可有效缩短分析检测时间,提高工作效率。通过方法学验证,证明该法具有准确快速、重复性好且杂质干扰小等特点。经3 类人参加工品生晒参、红参和黑参共15 批实际样品的检测分析验证,完全能够满足检测需求,可应用于实际人参加工品中皂苷类化合物的直接测定。
猜你喜欢
基层中医药(2021年7期)2021-11-02 07:20:06
海峡姐妹(2019年8期)2019-09-03 01:01:04
中成药(2018年12期)2018-12-29 12:26:08
中成药(2018年9期)2018-10-09 07:19:04
中成药(2017年9期)2017-12-19 13:34:40
中成药(2017年6期)2017-06-13 07:30:34
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:36:55
广东第二课堂·小学(2016年11期)2016-12-06 14:28:02
华人时刊(2016年13期)2016-04-05 05:50:15
合成化学(2015年1期)2016-01-17 08:55:47
