
UPLC法测定硫酸羟氯喹原料药有关物质
2023-10-21 07:38伍良涌赵雪梅李玮玲董顺玲
广东药科大学学报 2023年5期
伍良涌,赵雪梅,李玮玲,董顺玲
(广州市药品检验所,广东 广州 510160)
硫酸羟氯喹(hydroxychloroquine sulfate)化学名为2-({4-[(7-氯喹啉-4-基)氨基]戊基}-乙胺基)乙醇硫酸盐,最初用于疟疾治疗,上市后主要用于治疗类风湿性关节炎和红斑狼疮[1-4]。目前,已有证据表明羟氯喹对于多种病毒均有抑制作用[5-6]。硫酸羟氯喹在临床使用中可能发生不良反应,主要包括皮肤、消化系统、神经系统、视觉及心血管系统损害等[7]。
硫酸羟氯喹国内质量标准有卫生部药品标准(1992)、国家药品标准YBH 4 份(1、2、3、4)等,国外药典中欧洲药典EP11.0、英国药典BP2022 和美国药典USP-NF2021 均有收载[8-10]。其中卫生部药品标准(1992)和国家药品标准YBH 1 的有关物质检查方法为薄层色谱(TLC)法,方法复杂、费时且不能对杂质进行定量分析;国家药品标准YBH 2-4 的有关物质检查方法均为高效液相色谱(HPLC)法,但方法不一致且控制限度不统一,其中国家药品标准YBH4 建立了2 个液相系统进行有关物质检查。欧洲药典EP11.0、英国药典BP2022 和美国药典USPNF2021 收载有关物质检查项为UPLC 法,其中前两者方法完全相同,明确了7 个已知杂质分别为杂质A、B、C、D、E、F和G。
国家药品监督管理局网站数据显示,硫酸羟氯喹原料药国内有6家生产企业涉及6个批准文号[11]。本次研究收集了3家企业(1、2、3)的样品,各个企业需控制的杂质不尽相同,3 家企业分别明确并提供了各自需要控制的杂质对照品(企业工作对照品),全部杂质对照品仅用于色谱峰定位,其中企业1 提供了欧洲药典EP 列出的7 个杂质对照品和企业自命名杂质G′、杂质H、杂质I、杂质J 和杂质K;企业2提供的资料说明与主峰相对保留时间0.6 的杂质为欧洲药典EP 的杂质C;企业3 提供了自命名杂质:杂质1、杂质2、杂质3、杂质4、杂质5、杂质8、杂质9、杂质10和杂质11(未提供杂质6和杂质7,仅提供结构式且说明其工艺不产生)。
目前,硫酸羟氯喹及其制剂检测多为HPLC法[12-14]。本文按罗马数字Ⅰ-Ⅻ对各已知杂质命名(详见表1),参照欧洲药典EP的方法测定12个已知杂质,结果不能得到完全分离,经方法优化、筛选色谱条件的基础上建立的UPLC 法,可以一次性有效分离各企业提供的12 个已知杂质,专属性强、灵敏度高,统一了现有国内质量标准中有关物质检查方法,为有效控制硫酸羟氯喹中有关物质提供了参考。
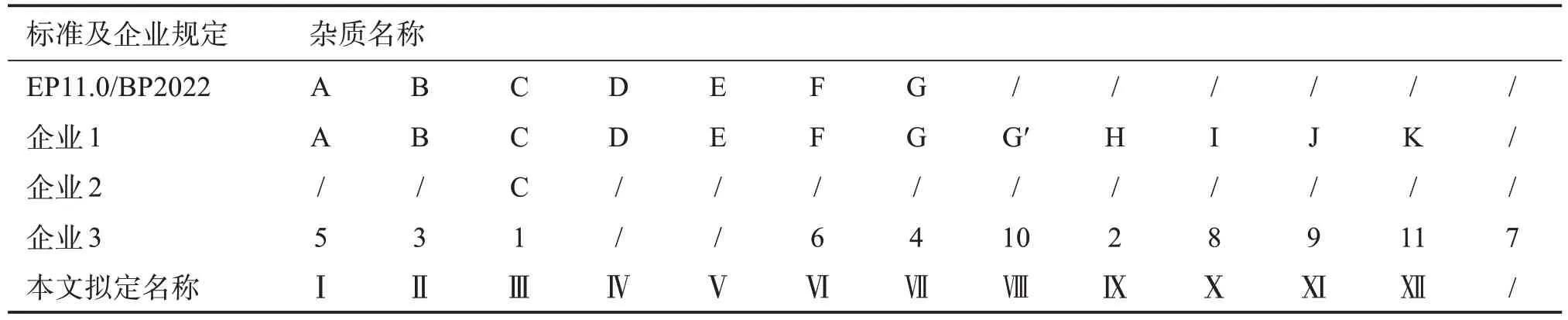
表1 硫酸羟氯喹原料中已知杂质比对Table 1 Comparison of known impurities in hydroxychloroquine sulfate API
1 仪器与试剂
Waters ACQUITY UPLC 超高效液相色谱仪,Mettler Toledo XSE205 DU 电子天平,Mettler Toledo S20K pH计。
庚烷磺酸钠(CNW,分析纯)和KH2PO4(广州市化学试剂厂,化学纯);甲醇(Merck,色谱纯);超纯水(Milli-QReferenceA+);硫酸羟氯喹对照品(中国食品药品检定研究院,批号100582-201902,质量分数99.3%),其他杂质对照品均为企业工作对照品;硫酸羟氯喹样品:企业1(批号:HS20171125,HS20171126,HS20171127,HS20181101V,HS2018110 2V,HS20181103V),企业2(批号:C40-170301,C40-170302,C40-170303),企业3(批号:SQK180407,SQK180408,SQK180409)。
2 方法与结果
2.1 色谱条件
参考欧洲药典EP 等国外药典[8-10]并结合企业意见,通过调整流动相中庚烷磺酸钠的浓度、梯度比例和色谱参数建立:以Waters ACQUITY UPLC®BEH C18(2.1 mm×150 mm,1.7 μm)为色谱柱,以磷酸盐缓冲液(取庚烷磺酸钠0.25 g 和KH2PO41.36 g,加水900 mL 溶解,用三乙胺调节pH值7.0,加水稀释至1 000 mL,摇匀)-甲醇(体积比90∶10)为流动相A,以磷酸盐缓冲液-甲醇(体积比15∶85)作为流动相B,梯度洗脱(0~1 min,100%A;1~18 min,100%→20%A;18~20 min,20%A;20~25 min,20%→100%A);流速为0.25 mL/min;柱温为45 ℃;检测波长为254 nm;进样体积为4 μL。
2.2 溶液的配制
2.2.1 系统适用性溶液 取硫酸羟氯喹、杂质B(杂质Ⅱ)和杂质C(杂质Ⅲ)对照品适量,加溶剂[甲醇-水-10%硫酸溶液(体积比50∶50∶0.4),下同]超声溶解并稀释制成每1 mL中约含硫酸羟氯喹0.5 mg、杂质B 10 μg和杂质C 10 μg的混合溶液。
2.2.2 对照品溶液的配制 取硫酸羟氯喹对照品适量,精密称定,加溶剂溶解并定量稀释制成每1 mL中约含1 μg的溶液。
2.2.3 杂质对照品溶液 取杂质B、杂质C 对照品适量,分别加溶剂超声溶解并稀释制成每1 mL中约含杂质B 10 μg、杂质C 10 μg的杂质对照品储备溶液;取上述杂质对照品储备溶液适量,分别加溶剂稀释制成每1 mL中约含杂质B 1 μg、杂质C 1 μg的溶液。
2.2.4 混合对照品溶液 取硫酸羟氯喹、杂质A(杂质Ⅰ)至杂质11(杂质Ⅻ)共12个企业工作对照品适量,加溶剂超声溶解并稀释制成每1 mL中约含硫酸羟氯喹0.5 mg、杂质A(杂质Ⅰ)至杂质11(杂质Ⅻ)分别10 μg的混合溶液。
2.2.5 供试品溶液的配制 取硫酸羟氯喹样品适量,精密称定,加溶剂溶解并稀释制成每1 mL 中约含0.5 mg的溶液。
2.2.6 灵敏度溶液的配制 精密量取对照品溶液5 mL,置20 mL量瓶中,用溶剂稀释至刻度,摇匀。
2.3 测定方法
精密量取系统适用性溶液、混合对照品溶液、灵敏度溶液、供试品溶液与对照品溶液,分别注入超高效液相色谱仪,记录色谱图。杂质计算方法及限度均参考欧洲药典EP11.0 和英国药典BP2022,采用加校正因子的主成分外标法计算。
2.4 方法学考察
2.4.1 系统适用性试验 系统适用性溶液色谱图中,硫酸羟氯喹峰的保留时间约为16 min,杂质B与杂质C、杂质C 与硫酸羟氯喹峰之间的分离度均应不小于3.0。灵敏度溶液色谱图中,硫酸羟氯喹峰的信噪比应大于10。系统适用性试验色谱图见图1。杂质B 与杂质C 色谱峰之间的分离度为3.0,杂质C与硫酸羟氯喹色谱峰之间的分离度为13.2;混合对照品溶液色谱图见图2,供试品溶液色谱图见图3,各杂质峰相对主峰的保留时间见表2。

图1 系统适用性试验色谱图Figure 1 Chromatogram of system suitability test
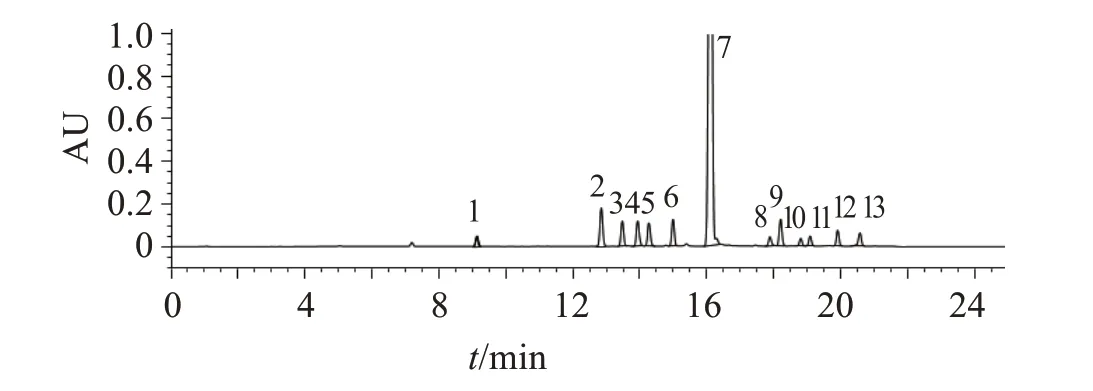
图2 混合对照品溶液色谱图Figure 2 Chromatogram of mixed reference solution
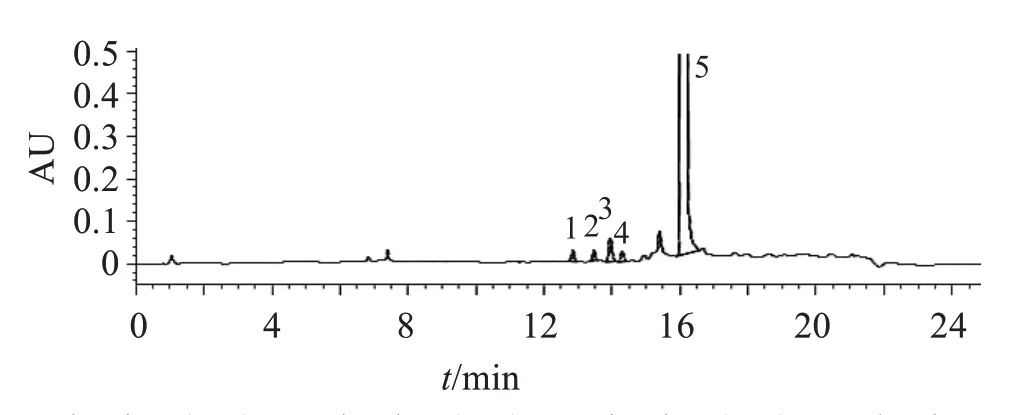
图3 有关物质供试品溶液色谱图Figure 3 Chromatogram of related substances in sample solution
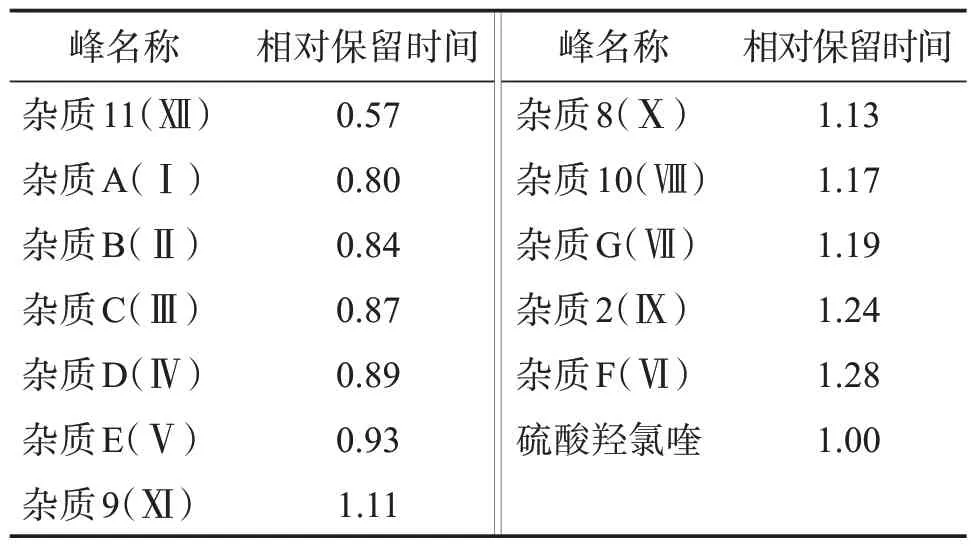
表2 各杂质相对保留时间Table 2 Relative retention time of related substances
2.4.2 专属性试验 取硫酸羟氯喹(企业1,批号HS20181101V)适量,进行酸破坏(2 mol/L HCl 溶液、水浴加热30 min)、碱破坏(2 mol/L NaOH 溶液、水浴加热30 min)、氧化破坏(30%的双氧水、水浴加热30 min)、热破坏(水浴加热30 min)、光照破坏(365 nm 照射60 min),并用溶剂稀释成一系列硫酸羟氯喹破坏性溶液(质量浓度为0.5 mg/mL)。结果显示,硫酸羟氯喹在酸性、碱性、氧化、热及UV 破坏条件下相对比较稳定,仅在365 nm 紫外光照破坏下杂质B增加,其余破坏方式产生的未知杂质较少,产生的未知杂质和各已知杂质与主峰分离良好,不干扰测定,详见图4。

图4 有关物质专属性试验色谱图Figure 4 Chromatogram of related substances in specificity test
2.4.3 线性关系考察 配制系列质量浓度为0.05~5.00 μg/mL(n=9)的硫酸羟氯喹对照品溶液,分别精密量取4µL,注入超高效液相色谱仪,记录色谱图,测定峰面积。以质量浓度(C)为横坐标、峰面积(A)为纵坐标绘制标准曲线,得线性方程A=26 191.71C+915.20,r=0.999 9,表明硫酸羟氯喹在0.05~5.00 μg/mL质量浓度范围内与峰面积线性关系良好。
2.4.4 精密度试验 取对照品溶液连续测定7次,测得硫酸羟氯喹峰面积的RSD值为0.4%(n=7)。
2.4.5 检测限和定量限 取硫酸羟氯喹对照品溶液,逐步稀释、进样,按信噪比3∶1计算检测限、10∶1计算定量限,结果分别为0.18 ng和0.60 ng。
2.4.6 稳定性试验 取供试品溶液(批号:HS2017 1125、C40-170301 和SQK180407)室温放置,分别于0、9、13、18 h进样测定。结果显示,杂质个数无显著变化,单个杂质和杂质总量基本稳定,其中批号为HS20171125 的供试品溶液中杂质B(Ⅱ)、杂质C(Ⅲ)和杂质总量的峰面积RSD 值分别为0.7%、3.3%、2.4%(n=4),批号为C40-170301 的供试品溶液的分别为“未测出”、4.3%、6.2%(n=4),批号为SQK180407 的供试品溶液的分别为2.1%、7.2%、7.0%(n=4),表明供试品溶液在18 h内基本稳定。
2.4.7 重复性试验 取硫酸羟氯喹样品(批号:HS20171125、C40-170301 和SQK180407)照“2.2.5”方法制备供试品溶液6 份,按“2.1”项色谱条件进样测定,结果显示批号为HS20171125 的供试品溶液中杂质B(Ⅱ)、杂质C(Ⅲ)和杂质总量的含量RSD值分别3.6%、3.8%、3.3%(n=6),批号为C40-170301的供试品溶液的分别为“未测出”、3.7%、5.0%(n=6),批号为SQK180407 的供试品溶液的分别为7.5%、10.2%、10.7%(n=6),表明方法重复性良好。
2.4.8 回收率试验 取约50 mg硫酸羟氯喹样品(批号:SQK180407),精密称定,置100 mL容量中,制备6 份,分别加入“2.2.3”项下质量浓度为10 μg/mL 杂质B和杂质C对照品储备溶液10 mL,稀释至刻度。精密量取“2.2.3”项下杂质对照品溶液、上述6份溶液各4 μL,注入超高效液相色谱仪,记录色谱图和计算回收率。结果显示,杂质B 的回收率在100.7%~104.3% 范围内,平均回收率为102.2%,RSD 为1.6%;杂质C的回收率在100.1%~101.8%范围内,平均回收率为100.8%,RSD 为0.7%,表明方法准确度良好。
2.4.9 有关物质测定结果 12 批样品测定结果,见表3。可见,企业1 提供6 批样品中前3 批样品结果与后3 批结果有差别,后3 批样品结果与企业2、企业3提供样品结果比较相近,后9批样品中杂质B均<0.1%,杂质C 均<0.2%,其他单个杂质均<0.1%,杂质总量均<0.25%。
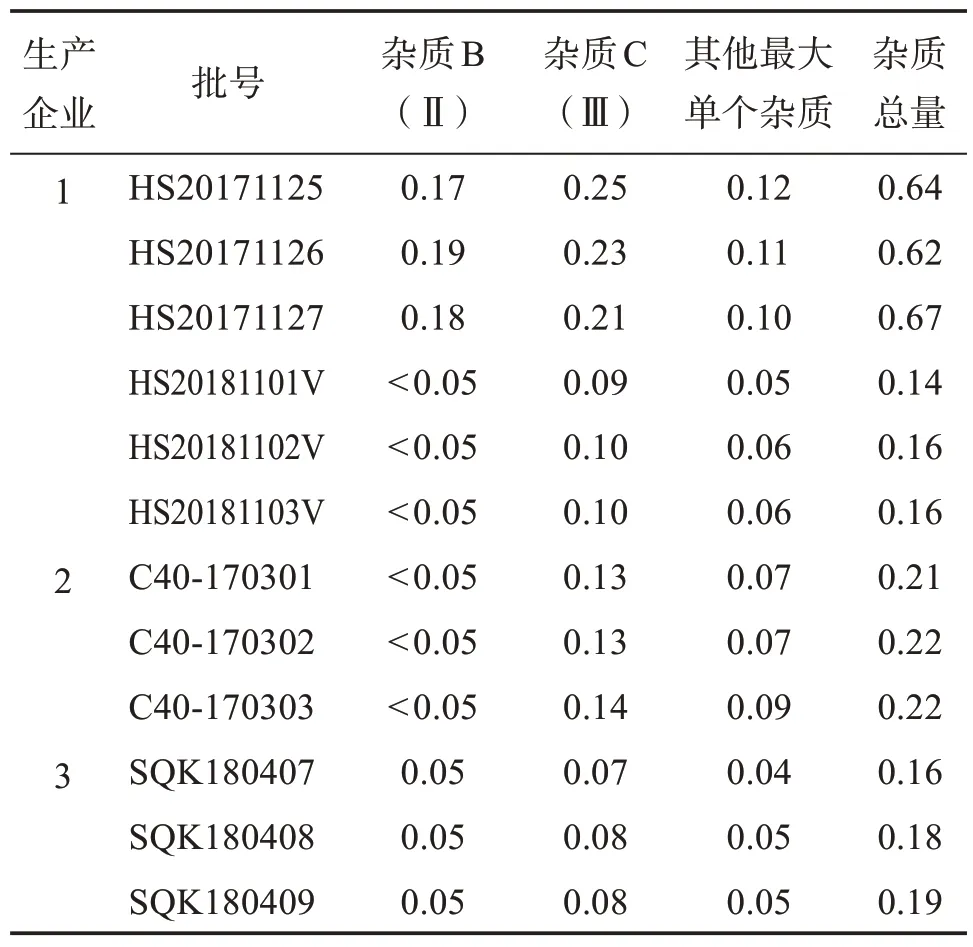
表3 有关物质测定结果Table 3 Results of determination of related substance w/%
2.4.10 拟定杂质的限度 采用企业提供的杂质对照品定位,计算各已知杂质的相对保留时间(见表2),杂质计算方法及限度均参考欧洲药典EP11.0 和英国药典BP2022,采用加校正因子的主成分外标法计算。结果测得,拟定限度为杂质B(乘以校正因子1.6)不得过0.15%,杂质C 不得过0.4%,其他单个杂质不得过0.10%,杂质总量不得过0.6%。小于灵敏度溶液主峰面积的杂质峰忽略不计(0.05%)。
3 讨论
3.1 硫酸羟氯喹及主要杂质的结构
杂质B(Ⅱ)、杂质C(Ⅲ)和硫酸羟氯喹的结构(图5)类似,光谱图(图6)也基本相同,故本研究选择杂质B 与杂质C、杂质C 与羟氯喹峰之间的分离度定为方法系统适用性要求。
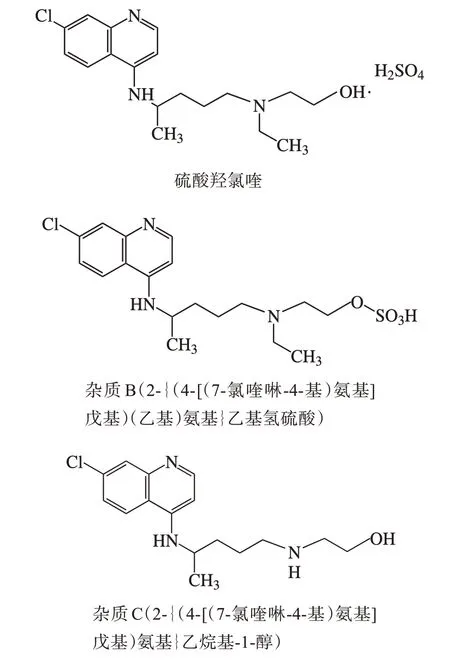
图5 杂质结构式Figure 5 Formula of impurities
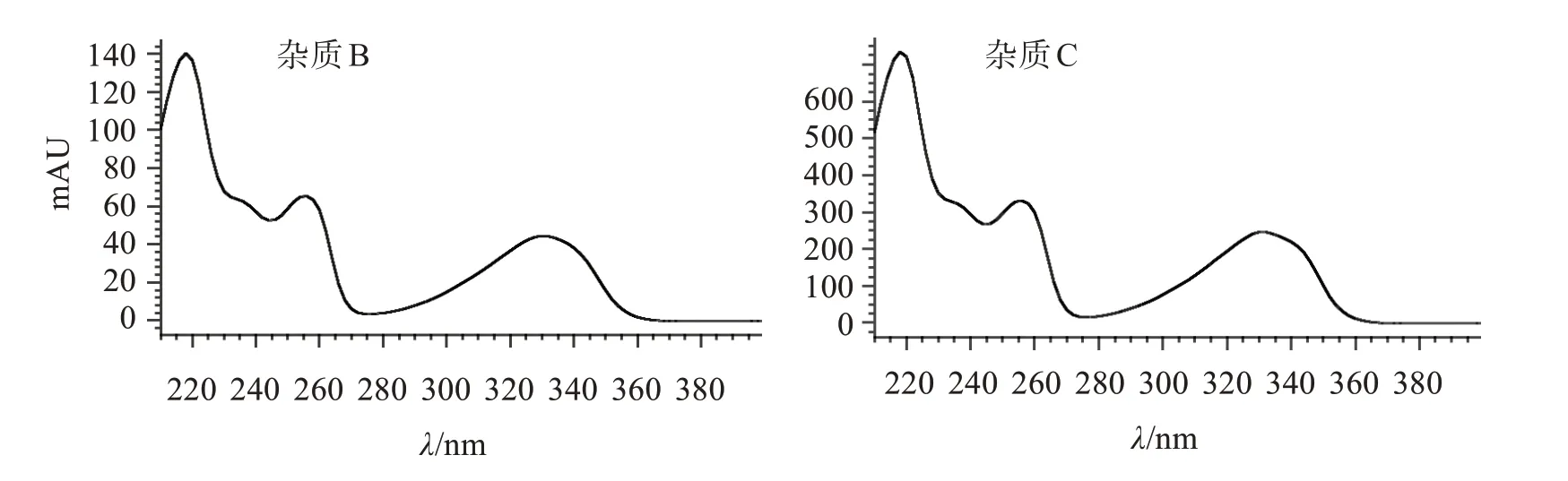
图6 硫酸羟氯喹杂质B和杂质C光谱图Figure 6 Spectrogram of impurity B and impurity C of hydroxychloroquine sulfate
3.2 检测波长的选择
欧洲药典EP11.0/英国药典BP2022和YBH 2标准,分别采用波长254 nm 和220 nm 测定本品的有关物质,结果表明采用220 nm 检测基线漂移较大,空白干扰较大(见表4),最终选择检测波长254 nm。
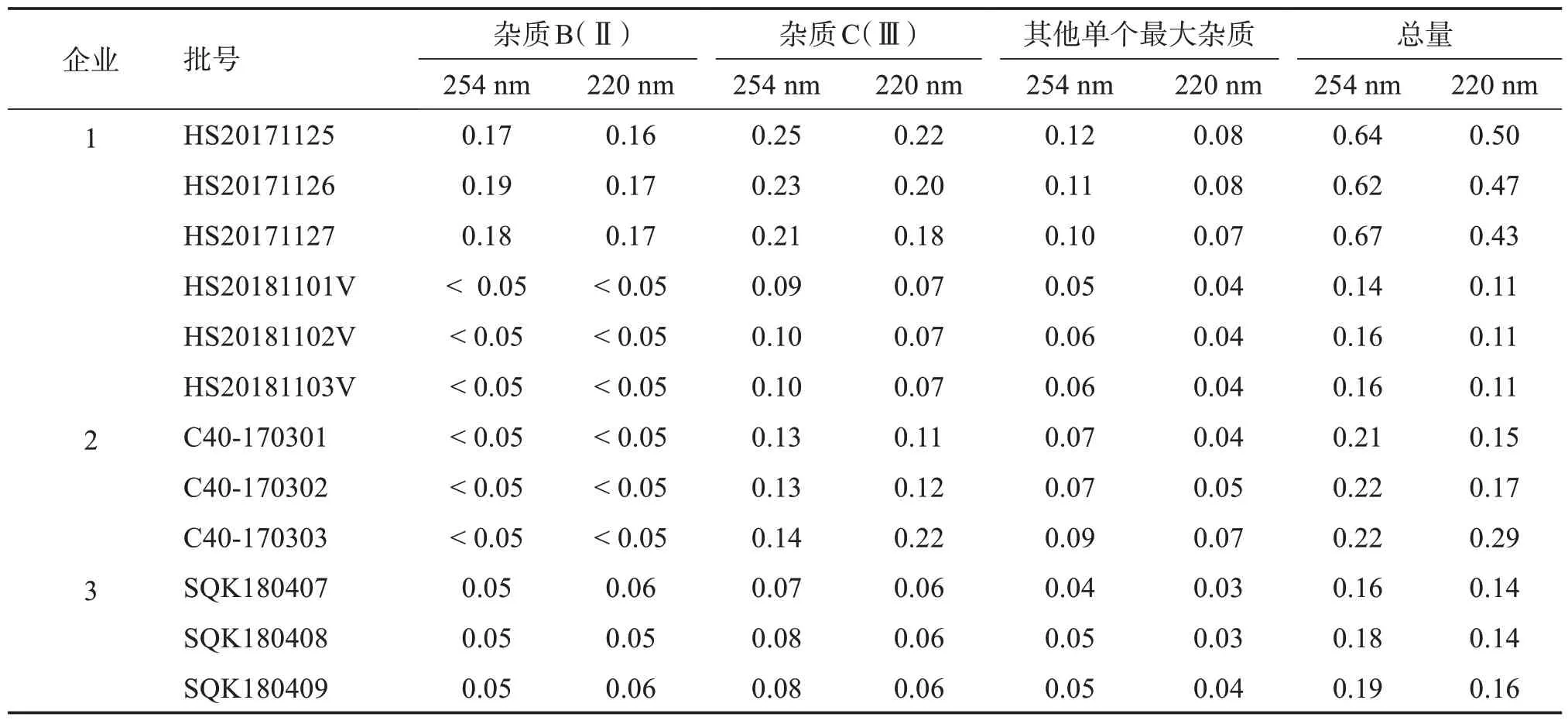
表4 2个波长各杂质结果比对表Table 4 Comparison results of two wavelengths for related substancesw/%
3.3 色谱条件优化
在实验过程中发现,采用不同品牌的庚烷磺酸钠对基线有一定的影响,分别比较了庚烷磺酸钠(2个不同批号)和Fisher Chemical的庚烷磺酸钠,结果同一品牌不同批号或者不同品牌的庚烷磺酸钠造成的波动大小不一;色谱柱多次进样后,在主峰保留时间处会有主成分残留;色谱柱多次进样后或者庚烷磺酸钠质量不佳时,基线噪音增大,出现鬼峰。若出现以上现象,建议可采取用10%甲醇溶液和甲醇交替多次冲洗色谱柱,在液相色谱仪混合器之后进样器之前安装鬼峰捕集小柱(Nano ChromGhodt-Remover, 2.1 mm×50 mm)减少或消除波动及噪音的影响,更换色谱柱等方法解决。
4 结语
本文参考欧洲药典方法,结合征集生产企业的合成工艺和已知杂质,整理出12个已知杂质并重新排序命名,通过适当调整流动相的比例及修改洗脱梯度建立了UPLC 法,有效分离并准确测定硫酸羟氯喹原料药的有关物质,为硫酸羟氯喹质量控制及生产企业改进工艺提供了参考。
本研究采用加校正因子的主成分外标法计算有关物质,对硫酸羟氯喹进行了线性关系、精密度、检测限和定量限方法学考察。选择杂质B、杂质C和杂质总量为关键评估指标对供试品进行了稳定性、重复性和回收率考察,其中供试品的主要杂质和杂质总量因测定结果偏小,故稳定性和重复性RSD 偏大,但对比实际测试结果偏差均在可接受范围内。本研究未能获取商业化硫酸羟氯喹杂质对照品,12 个杂质对照品均为企业自制工作对照品纯度不高且获得量少,有待今后改进。
(利益冲突声明:本研究未受到企业、公司资助,不存在利益冲突。)
猜你喜欢
实用药物与临床(2021年8期)2021-09-14
中国药理学与毒理学杂志(2020年4期)2020-08-14
实用心脑肺血管病杂志(2020年3期)2020-05-26
广州化工(2020年5期)2020-03-31
浙江大学学报(工学版)(2016年11期)2016-06-05
中国猪业(2016年1期)2016-01-28
合成化学(2015年9期)2016-01-17
化工进展(2015年6期)2015-11-13
中国中医药现代远程教育(2014年11期)2014-08-08
郑州大学学报(工学版)(2014年6期)2014-03-01
